
Abstract
Background
Progressive supranuclear palsy (PSP) is a rare neurodegenerative disease with diverse clinical phenotypes, prompting the development of new diagnostic criteria known as the MDS-PSP classification. However, little is known about the prognostic value of this classification in order to better stratify patients for the clinical trials.
Objective
To assess the impact of the different clinical phenotypes according to the MDS-PSP classification on prognosis using the clinical milestones of death, severe dysphagia, institutionalization, and need for walking aid.
Methods
A prospective cohort of 205 PSP patients from Lille University Hospital was analyzed retrospectively. Patients were classified into different MSD-PSP phenotypes according to their clinical presentation after 3 years of follow-up. The milestones of death, severe dysphagia, institutionalization, and need for walking aid were recorded, and a survival analysis was performed to describe the prognosis of each disease presentation.
Results
Median survival time was 6.4 (interquartile range (IQR): 4.8–8.6) years and mean diagnostic delay from symptom onset was 38.1 ± 22.5 months. PSP Richardson Syndrome (PSP-RS) had a poorer survival rate and a higher occurrence of severe dysphagia and need for walking aid compared to PSP variants such as PSP Parkinsonism (PSP-P), PSP postural instability without ocular motor dysfunction (PSP-PI), and other rare phenotypes.
Conclusions
PSP-RS has a less favorable prognosis compared to PSP variants stratified according to the MDS-PSP classification. This classification could assist in selecting patients for clinical trials and help design outcomes that account for the disease heterogeneity.
Plain language summary
Progressive supranuclear palsy (PSP) is a rare and debilitating neurodegenerative disease marked by diverse combination of motor and cognitive symptoms. No treatment is currently available to slow down disease progression. The multiple clinical presentations of PSP compromise clinical trials. Thus, we aimed to study the differential prognosis of the different forms of the disease. We showed that PSP-Richardson Syndrome (PSP-RS), which is the most frequent presentation, seems to have a poorer prognosis than other variants on survival and other disability criteria such as severe dysphagia occurrence. This study suggests that these differences should account to improve the design of future clinical trials.
Keywords
Introduction
Progressive supranuclear palsy (PSP) is a rare, sporadic, neurodegenerative disease characterized by a combination of parkinsonism, ocular gaze palsy, early postural disorders, falls, pseudobulbar syndrome, and cognitive impairment. However, the expression of these symptoms varies in intensity and temporality, leading to different clinical phenotypes.1,2 PSP is extremely severe and rapidly compromises autonomy, quality of life, and survival in just a few years without any specific treatment.
Clinical criteria were developed in order to standardized ante-mortem diagnosis 3 and to better describe the phenotypic heterogeneity of PSP. 4
The most common phenotype, PSP-Richardson Syndrome (PSP-RS), was reported as a combination of vertical supranuclear palsy and falls occurring during the first year of the disease. Other phenotypes are characterized by predominant parkinsonism (PSP-P), cognitive or behavioral disorders (PSP-F), speech or language alterations (PSP-SL), cortico-basal syndrome (PSP-CBS), postural instability without ocular motor dysfunction (PSP-PI), progressive gait freezing (PSP-PGF), or predominant ocular motor dysfunction (PSP-OM). 4 These variants represent about half of PSP patients.5,6
PSP-RS seems to have a poor prognosis, with death occurring between 5–7 years after disease onset and 3–4 years after diagnosis,7–10 but little is known about the prognosis of other phenotypes of the disease. Indeed, only a few studies are available and most focus on comparing PSP-RS with non-PSP-RS, mainly based on the previous National Institute of Neurological Disorders Society for PSP (NINDS-SPSP) classification. 3
It is therefore essential to be aware of the prognosis of the different phenotypes in order to know whether they can be combined in a multicenter trial and, if not, how patients should be stratified for disease-modifying therapy trials.
Our aim was therefore to determine the relation between MDS-PSP classification and prognosis using four clinical endpoints: death, severe dysphagia, need for walking aid, and institutionalization.
Methods
Study population
Between January 2008 and February 2019, 292 consecutive patients were assessed for suspected PSP at the Movement Disorders Department or the Memory Center of Lille University Hospital. Their medical records were analyzed retrospectively, and each patient was assigned to a clinical MDS-PSP phenotype that best described them by an expert neurologist in movement disorders.
As recommended by the Movement Disorder Society, Multiple Allocations eXtinction (MAX) rules were applied to define the predominant phenotype, when more than one phenotype was appropriate for one subject. 11 If several phenotypes were still possible despite MAX rules, the phenotype was considered as undetermined and the patient was excluded from the analysis.
Patients meeting the MDS-PSP mandatory exclusion criteria, 4 with predominant episodic memory impairment suggestive of Alzheimer's disease (AD), with severe leukopathy or, when available, with a different neuropathologic diagnosis, were excluded from the statistical analysis.
Due to the small number of subjects and to the low number of events in these groups, PSP-PGF, PSP-F, PSP-SL, and PSP-CBS were grouped together as “Others” for the analysis. As in previous publications,12,13 non-PSP-RS was also analyzed by dividing the patients into PSP-cortical (including PSP-F, PSP-SL, and PSP-CBS) and PSP-subcortical (including PSP-P, PSP-PI, and PSP-PGF).
When available, pathologic validation of the diagnosis was considered as an inclusion or exclusion criterion.
Study outcome measures
The primary outcome measure of the study was death. Three other clinical milestones that have significant consequences on quality of life were also selected a priori, namely severe dysphagia, need for walking aid, and institutionalization. 7
Dysphagia was considered to be severe if it led to aspiration pneumonia, if it required adapted food texture, or if percutaneous endoscopic gastrostomy was necessary.
The patients’ medical records were then reviewed in the hospital database to document the onset and timing of pre-defined clinical milestones, from symptom onset to disease progression.
Ethics
All examinations were performed as part of routine care. Analyses were carried out retrospectively. After consultation with our ethics committee, no authorization or informed consent was required.
Statistical analysis
Categorical variables are expressed as frequency and percentage, and quantitative variables as mean ± standard deviation (SD) in the case of normal distribution, or as median (interquartile range, IQR) otherwise. Normality of distribution was checked graphically and using the Shapiro-Wilk test. Patient survival was estimated by the Kaplan-Meier method and compared between PSP groups using a Cox proportional hazard model adjusted for predefined confounding factors (sex and age). Hazard-ratios (HRs) and their 95% confidence intervals (95%CI) were derived from models as effect sizes. Cumulative incidence of need for walking aid, cumulative incidence of severe dysphagia, and cumulative incidence of institutionalization were estimated by the Kalbfleisch and Prentice method considering death as a competing event and compared between PSP groups using a Fine and Gray competing risk regression model adjusted for predefined confounding factors. Sub-HRs and their 95%CI were derived from models as effect sizes. Statistical testing was conducted at the two-tailed α-level of 0.05. Data were analyzed using the SAS software version 9.4 (SAS Institute, Cary, NC).
Results
Study population
A total of 292 patients were assessed for suspected PSP and 205 were diagnosed with clinically diagnosed PSP according to MDS-PSP criteria.
Ninety-six patients were excluded from the statistical analysis: 67 because of a more likely differential diagnosis (11 Parkinson's disease, 10 multiple system atrophy, nine corticobasal syndrome (CBS), four normal pressure hydrocephalus, three Lewy body dementia, three AD, two amyotrophic lateral sclerosis, one vascular parkinsonism, and 24 other neurodegenerative disease), four because of predominant episodic memory impairment suggestive of AD, three because of severe leukoencephalopathy, 11 because of missing data, and two because of a pathologic diagnosis of corticobasal degeneration (CBD). Finally, nine patients with clinically diagnosed PSP were excluded from the survival analysis because they were classified as undetermined PSP phenotype. Thus, 196 were enrolled for the survival analysis.
A total of 102 (49.8%) patients were classified as PSP-RS, 31 (15.1%) as PSP-PI, 28 (13.7%) as PSP-P, 14 (6.8%) as PSP-SL, 10 (4.9%) as PSP-F, seven (3.4%) as PSP-PGF, four (2%) as PSP-CBS, and nine (4.4%) as undetermined phenotype (Figure 1).
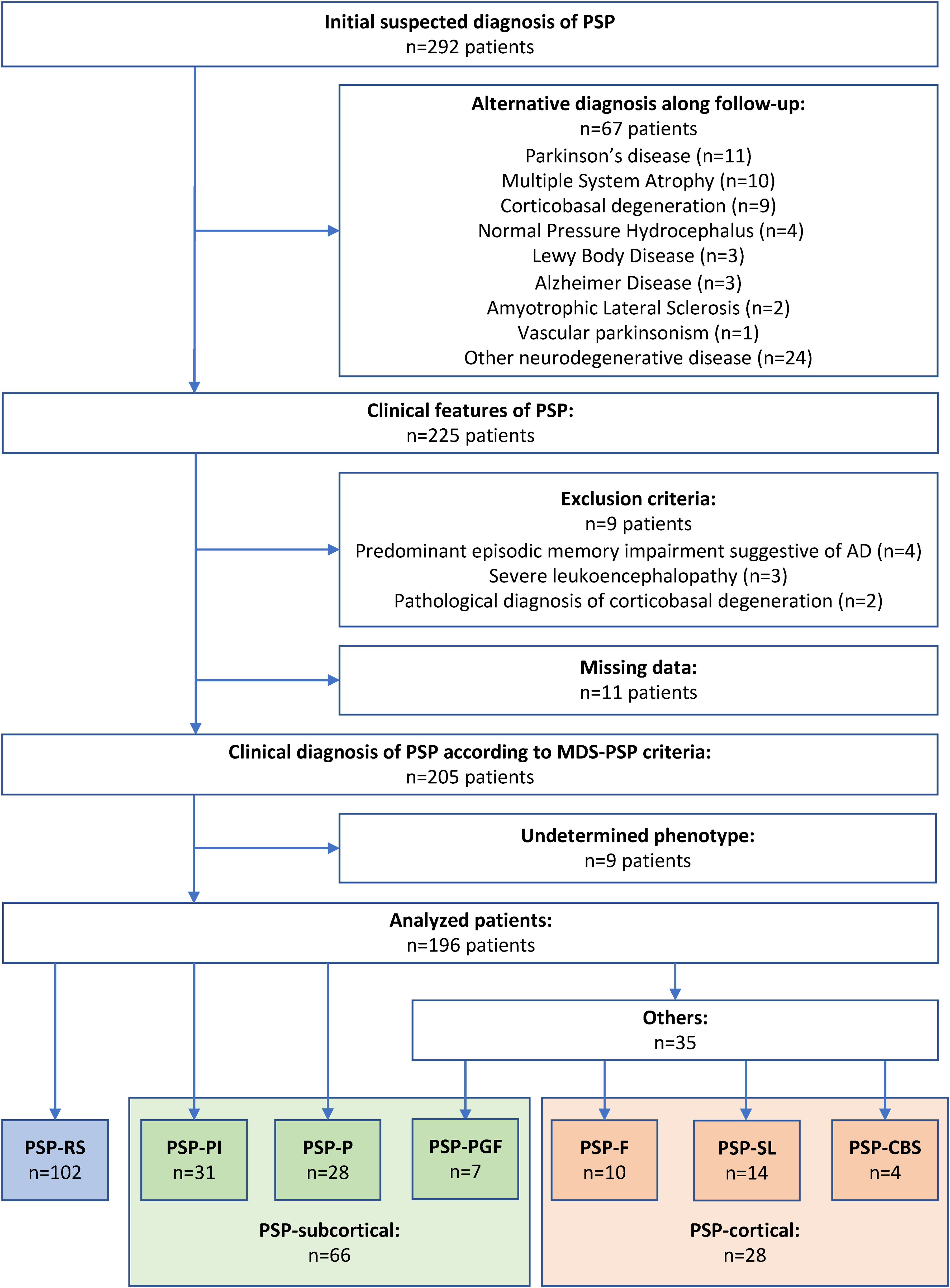
Flow diagram for the study population.
The pathologic diagnosis of PSP was confirmed a posteriori for six patients.
The sex ratio (M/F) was 1.23 and mean (SD) age at disease onset was 67.6 ± 7.5 years. Survival rate at 10 years was 12.4% [95%CI: 7.5–18.6]. Median survival time was 6.4 (IQR: 4.8–8.6) years and mean diagnostic delay from symptoms onset was 38.1 ± 22.5 months. For the 42 living patients at the end of follow-up, median follow-up duration was 6.1 (IQR: 4.7–8.0) years (Supplemental Table 1).
Comparisons between the different phenotypes
Cumulative incidence of milestones at 10 years was 52.1% for severe dysphagia, 73.0% for walking aid, and 19.5% for institutionalization (Figure 2, Supplemental Table 2, and Supplemental Figure 1).
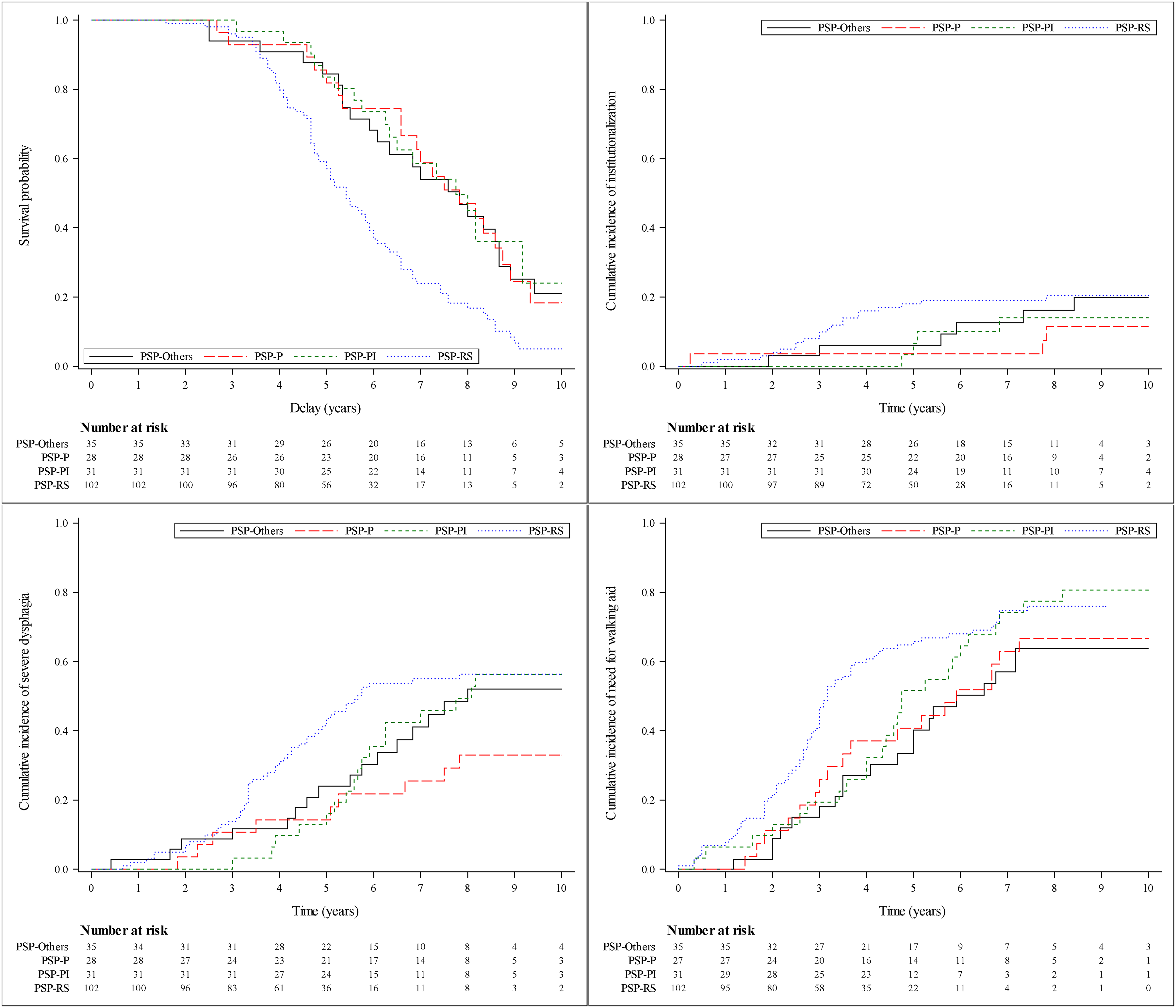
Survival and milestones cumulative incidence curves according to phenotypes groups.
All non-PSP-RS categories (PSP-P, PSP-PI, and others) had a significantly lower risk of death than PSP-RS (HR = 0.42 [95%CI: 0.25–0.70], HR = 0.39 [95%CI: 0.23–0.65], and HR = 0.34 [95%CI: 0.20–0.56], respectively). Risk of severe dysphagia was significantly lower for PSP-P than PSP-RS (HR = 0.43 [95%CI: 0.21–0.87]) and need for walking aid was significantly lower for other PSP than PSP-RS (HR = 0.58 [95%CI: 0.36–0.93]). When comparing PSP-cortical and PSP-subcortical to PSP-RS, both had a significantly lower risk of death (respectively HR = 0.38 [95%CI: 0.22–0.65] and HR = 0.38 [95%CI: 0.25–0.56]), respectively. PSP-subcortical had a lower risk of severe dysphagia than PSP-RS, and PSP-cortical had a lower risk of need for walking aid than PSP-RS (Table 1).
Comparison of milestones occurrence between phenotypes groups.
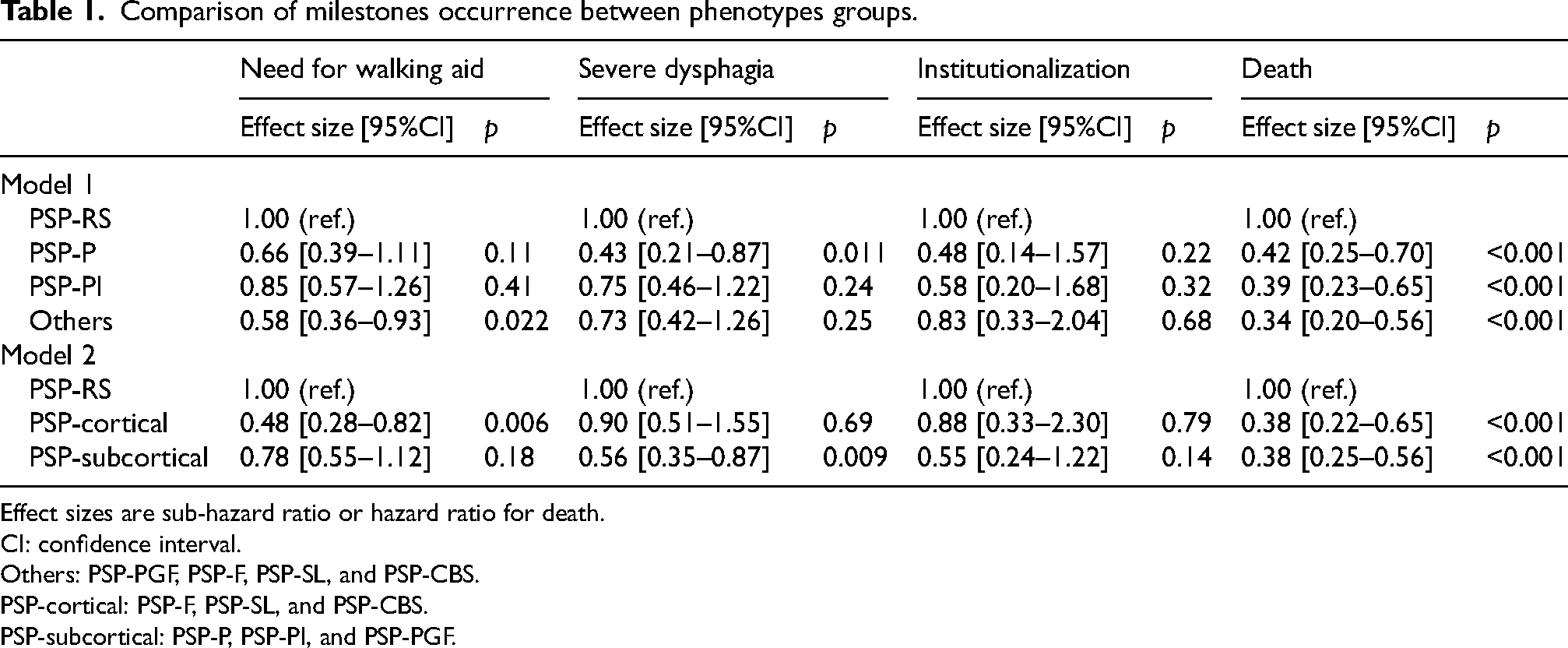
Effect sizes are sub-hazard ratio or hazard ratio for death.
CI: confidence interval.
Others: PSP-PGF, PSP-F, PSP-SL, and PSP-CBS.
PSP-cortical: PSP-F, PSP-SL, and PSP-CBS.
PSP-subcortical: PSP-P, PSP-PI, and PSP-PGF.
Discussion
This study confirms that PSP-RS is associated with a higher risk of death compared to all the other PSP phenotypes according to the MDS-PSP classification, with significantly longer life expectancy in patients with PSP-P, PSP-PI, and in a subgroup including other rarer phenotypes. Moreover, when PSP-cortical and PSP-subcortical were combined as non-PSP-RS patients, the results observed were consistent with previous reports.12,13
The present study also shows that patients with PSP-RS have a higher risk of severe dysphagia compared to the PSP-P and PSP-subcortical subgroups and a higher risk of needing a walking aid compared to other rare PSP and PSP-cortical subgroups. A large meta-analysis highlighted that early dysphagia, cognitive disorders, and falls were associated with a shorter life expectancy in PSP. 9 Another clinic-pathologic study reported as well a longer delay before dysphagia occurrence in PSP-P than in other phenotypes. 14 These previous publications also reported that PSP-RS was an unfavorable predictor of survival compared to PSP-P.9,14
In contrast, there was no influence of PSP phenotype on institutionalization, suggesting the MDS-PSP classification did not influence this factor; this milestone is influenced by factors other than disease progression, such as the socio-economic and family environment.
To our knowledge, this is the first prognostic stratification based on the MDS-PSP classification. None of the previously published studies detailed the prognostic value of this classification for each MDS-PSP phenotype, but mainly focused on comparing PSP-RS with non-PSP-RS or with PSP-cortical and PSP-subcortical subgroups. Our study clearly establishes, for the first time, that patients with PSP-RS have a higher risk of severe dysphagia than those with PSP-subcortical, and a greater need of a walking aid compared to PSP-cortical and other rare PSP phenotypes.
Our results are consistent with previous studies, with a shorter survival in PSP-RS than in variant phenotypes,9,12–14,15 which can have huge implications for patient selection and outcome definition in clinical trials.
In a small sample study on 32 patients, 12 the authors reported a significantly shorter survival in PSP-RS compared to non-PSP-RS. In this publication, time to death was even shorter when combining PSP-RS with PSP-cortical compared to PSP-subcortical, whereas survival of cortical phenotypes was similar to PSP-RS. 12 Our larger cohort analysis showed that PSP-cortical also had a significantly better prognosis in terms of death when compared to PSP-RS. However, in a more recent study about 117 PSP patients, PSP-cortical was fund to present higher disability than PSP-subcortical according to motor, quality of life and neuropsychiatric scales, whereas PSP-subcortical survived longer than PSP-RS and PSP-cortical. 16 A similar size monocentric cohort of 227 patients, 13 also found coherent results, with longer survival in variant phenotypes than in PSP-RS. These authors also reported a faster rate of progression of cognitive deterioration in PSP-RS, but they did not analyze each phenotype in the MDS-PSP classification and failed to show a statistical difference on motor assessment. 13 A large retrospective cohort analysis of 334 patients aimed to determine the impact of the disease on daily living activities for each MDS-PSP subtype. 17 The authors reported that PSP-RS and PSP-PGF had a higher frequency of wheelchair dependency and that PSP-RS reached this milestone earlier than other subtypes. They also showed that PSP-P reached the milestones of wheelchair dependency, unintelligible speech, and severe dysphagia requiring percutaneous endoscopic gastrostomy later than other subtypes. The authors also reported that patients with PSP-F developed severe cognitive impairment earlier than others and reached this milestone earlier than those with PSP-RS. 17 This study corroborates our results with an earlier occurrence of severe dysphagia in PSP-RS than in PSP-P. We also found consistent results in our subgroup of patients with rare ‘other’ phenotypes, with longer survival without the need for a walking aid than in PSP-RS, whereas no difference was observed between the PSP-P and PSP-PI subgroups.
The present study has the strength of including one of the largest longitudinal PSP cohorts, with very few missing data. However, the study has also some limitations, notably the lack of a description for each rare MDS-PSP phenotype (PSP-PGF, PSP-F, PSP-SL, and PSP-CBS), because there were too few subjects or events in these groups. The retrospective analysis of a prospective cohort may have induced some biases. Notably, patient selection was based on the NINDS-SPSP criteria, and MDS-PSP criteria were applied retrospectively, which could have resulted in an underestimation of the prevalence of the other variants of the disease and of the prevalence of the specific milestones, especially for the patients with more severe disease progression. As well, patients who met the criteria for PSP-PI could only have a diagnostic certainty of “suggestive PSP”, making the diagnosis of PSP less certain and therefore, might have a different prognosis with underestimation of milestones occurrence. Data were collected from two units in a single expert center for movement disorders and our results should be confirmed in a broader range of patients from other centers. Finally, pathologic confirmation of the diagnosis was only available for six patients out of the 205, leading to potential inclusion of non-definite PSP patients.
Therapeutic clinical trials in PSP have recently been completed,18,19 and more are planned for the few years. However, approximately half of the PSP population do not meet the restrictive eligibility criteria for these trials. 13 These patients are therefore not well represented in trial cohorts and the differences between the phenotypes are not taken into account. Most of the previous clinical trials focused on PSP-Rating Scale 20 as the primary outcome, but this strategy is mostly biased by the differential progression profile between PSP-RS and non-PSP-RS. Some previous data also suggest a differential progression profile according to PSP-Rating Scale, with a faster progression of PSP-RS than PSP-P, even with similar severity at baseline. 21 In the same way, if a large phase 3 trial was conducted, even with a strong primary criteria as death occurrence, the phenotypic heterogeneity should biased the conclusion. To avoid this limitation, future clinical trials should consider the phenotypic variation in PSP by restricting patient selection to PSP-RS or non-PSP-RS phenotypes, or should stratify the population according to MDS-PSP classification. The same strategy should be applied if the outcome measure is the occurrence of severe dysphagia or need for a walking aid. Otherwise, the outcome criteria should be selected using a criterion which is not influenced by the MDS-PSP classification, such as institutionalization.
This study confirms that PSP variants have a different progression profile to PSP-RS, with a more favorable prognosis. Understanding the differential progression of the various clinical phenotypes of PSP is crucial to be able to accurately stratify patients as a function of prognosis to improve future clinical trial design.
Supplemental Material
sj-docx-1-pkn-10.1177_1877718X241291996 - Supplemental material for Clinical prognostic factors in progressive supranuclear palsy: Implications for clinical trials
Supplemental material, sj-docx-1-pkn-10.1177_1877718X241291996 for Clinical prognostic factors in progressive supranuclear palsy: Implications for clinical trials by Félix Marchand, Anne-Sophie Blaise, Luc Defebvre, Emeline Cailliau, Stéphanie Bombois, David Devos and Caroline Moreau in Journal of Parkinson's Disease
Footnotes
Acknowledgments
The authors thank the patients who took part in this study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by the INSERM UMRS-1172 research unit.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Luc Defebvre served on the Scientific Advisory Board for Abbvie and has received honoraria from pharmaceutical companies for consultancy and lectures including Abbvie, Novartis, Aguettant, and Orkyn.
Stéphanie Bombois received honoraria from Biogen for a symposium (2021) and from Esai for a lecture (2020), and is an investigator without personal fees in therapeutical trials for Alzheimer's disease from Biogen, Roche, Eisai, Eli Lilly, Janssen, Johnson & Johnson, Alector, NovoNordisk, Genentech, AB science, and Novartis.
David Devos has led four investigator-driven studies funded by academic grants from France from the French Ministry of Health and Research and the European Commission Horizon 2020 PHC13 2014–2015 (N° 633190), involving deferiprone, provided free of charge by ApoPharma Inc. and Chiesi Canada Corp. (FAIRPARK-I, FAIRPARK-II and SAFE-FAIR ALS-I, FAIR-ALS-II). He has served on advisory boards, served as a consultant and given lectures for pharmaceutical companies such as Orkyn, Abbvie, Medtronic, and Boston Scientific.
Caroline Moreau has received grants from the France Parkinson & Vaincre Parkinson charities, and French ANR. She has received various honoraria from pharmaceutical companies for consultancy and lectures on Parkinson's disease at symposia, including Orkyn, Elivie, Abbvie, and Boston Scientific. She is an Editorial Board member of this journal, but was not involved in the peer-review process of this article nor had access to any information regarding its peer review.
All other authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.