
Abstract
Background
Chiari II malformation occurs in one of 1000 live births and causes posterior fossa malformation. In Chiari II malformation, a lumbosacral meningomyelocele is nearly always present. Achondroplasia is the most common cause of dwarfism, occurring in one of 26,000 live births. Both Chiari II malformation and achondroplasia can cause compression at the craniocervical junction and consecutive hydrocephalus.
Case presentation
The case of a three-year-old male with Chiari II malformation, lumbosacral meningomyelocele, and achondroplasia is presented. To the authors’ knowledge, this is the second such case that has been reported so far. A surgical therapy of a lumbosacral meningomyelocele and an implantation of a ventriculoperitoneal shunt was performed in the first month after birth. At the age of two years, occipitoatlantoaxial stenosis required spinal decompression and laminectomy. The child presented in the outpatient department with life-threatening respiratory dysregulation, comprising prolonged expiratory apnoea with cyanosis (PEAC), acquired central hypoventilation syndrome, central sleep apnoea and obstructive sleep apnoea. He also presented with delayed language development, paraplegia, a neurogenic bladder, and dwarfism. The patient received non-invasive ventilation and had an individually adapted set of assistive and therapeutic devices. Cough insufficiency necessitated the adaption of mechanical insufflation-exsufflation. Speech and language therapy, physiotherapy, and occupational therapy were performed regularly. The patient started attending kindergarten just before his fourth birthday. At his one year follow-up, the patient's language capacities substantially improved and PEAC was not reported anymore.
Conclusion
When osseous, cerebral, and spinal disease are accompanied by life-threatening respiratory impairment, the following factors can reduce the impact of disability and can foster participation: treatment by an interdisciplinary team, the availability of assistive and rehabilitative technologies, living in a barrier-free home, a developmentally appropriate environment, and the continuous presence of trained caregivers.
Keywords
Introduction
Achondroplasia is the most common cause of disproportionate dwarfism and affects over 250,000 people worldwide. 1 It occurs in one of 26,000 live births and is caused by a mutation in the fibroblast growth factor receptor FGFR3 gene on chromosome 4p16.3. It encodes a tyrosine kinase receptor, which is expressed at different stages of development and plays a vital role in skeletal development. Due to those downstream effects, the conversion of cartilage to bone is impaired, particularly in the long bones of the arms and legs. Achondroplasia is inherited in the autosomal dominant manner with ∼80% of variants arising de novo. 2
Individuals with achondroplasia have a short stature, average-sized torso with short arms and thighs, and macrocephaly. Characteristic facial features are frontal bossing and midface retrusion. Furthermore, they develop a pronounced lordosis and kyphosis in childhood, adding to the prominent buttocks and protuberant abdomen. Their hands often have a trident configuration and limited elbow extension is typical. The acquisition of developmental motor milestones is often delayed, whereas intelligence and lifespan are usually close to normal. Health problems commonly associated with achondroplasia are obesity, recurrent ear infections due to the anatomy of the head, and spinal stenosis, typically in the lower back and less frequently in the foramen magnum. Hydrocephalus may be present, which can cause pressure on the brain stem with consecutive respiratory problems and increase the risk of death in infancy. Another respiratory complication of achondroplasia is obstructive sleep apnoea due to a combination of midface retrusion resulting in smaller upper airway size, hypertrophy of the lymphatic ring, airway malacia, and obesity. In extreme cases, adenotonsillectomy, positive airway pressure, weight reduction, or a tracheostomy can help. Ribcage deformity can also compromise respiratory function through elevated work of breathing. In the case of cervical cord compression at the craniocervical junction, a neurosurgical decompression or a ventriculoperitoneal shunt might be necessary. 3 Head circumference should be measured and neurological examination must be performed regularly. Additional routine MRI/CT imaging is recommended by some authors to check for hydrocephalus, as well as polysomnography to check for central sleep apnoea. Preventive strategies for corticomedullary compression include preventing sudden uncontrolled head movement, and avoiding umbrella strollers and baby carriers. 1
Guidelines on health supervision in achondroplasia were established by the American Academy of Pediatrics Committee on Genetics in 1995, and the last update was published in 2020. 4 These guidelines have facilitated the establishment of clinical cohorts to gather developmental data. A delay in development of gross motor and ambulatory skills and later communication abilities has been comprehensively described. 5 In terms of rehabilitation, a physically active lifestyle, moderate aerobic exercise, and strengthening training with or without weights <1 kg have been recommended to prevent or improve obesity, to optimize cardiopulmonary function, and to strengthen the skeletal muscles. Individuals with achondroplasia have a reduced vital capacity and lower tidal volume, resulting in a higher breathing frequency with lower ventilatory efficiency. Additionally, people with achondroplasia have a higher heart rate for a given oxygen uptake during exercise, implying that they may have a reduced cardiac stroke volume because of a smaller thoracic volume. Children with achondroplasia also have reduced muscle strength, supposedly due to abnormal musculoskeletal proportions and altered biomechanics. 6 These conditions may all be improved with physical exercise. 4 Additionally, environmental adaptations and activation of family resources can improve the health-related quality of life and functioning in young achondroplasia patients. 7 Families can benefit from anticipatory guidance and the opportunity to learn from other families with children of disproportionate short stature.
Chiari II malformation is a congenital malformation of the central nervous system. It is a malformation of the posterior fossa and spine leading to a beaked midbrain and downward displacement of the tonsils and cerebellar vermis through the foramen magnum, which can cause severe morbidity and mortality if not diagnosed and treated early. Chiari II malformation occurs in one of 1000 live births and is associated with neural tube defects like spinal myelomeningocele and hydrocephalus in more than 70% of patients. Even though the condition's pathogenesis mechanism is unknown, it has been shown that in animal models, open neural tube defects can cause a Chiari II malformation. One theory is that the cerebrospinal fluid leak through the neural tube defect can cause a drop in intracranial pressure. The pressure loss during foetal development can lead to poor cranial vault expansion, culminating in a small posterior fossa. The thus narrowed posterior fossa leads to downward displacement of the tonsils and cerebellar vermis through the foramen magnum. 2
The clinical presentation of Chiari II malformation varies according to severity, with arm weakness, aspiration, stridor, and other respiratory symptoms being the most common. Later in life, scoliosis or syrinx may occur. Sleep-disordered breathing occurs in approximately 45% of patients with Chiari II malformation and meningomyelocele, namely central sleep apnoea, obstructive sleep apnoea, and acquired central hypoventilation syndrome (presenting with hypercapnia, especially at night). 8 Vocal cord palsy has also been reported in patients with Chiari II malformation and meningomyelocele, 9 though it tends to improve as the patients get older. Prolonged expiratory apnoea with cyanosis (PEAC) is “continued expiratory activity at low lung volume with partial or complete glottic closure” associated with cyanosis. 10 The terminology is not uniform, and the terms “cyanotic expiratory apnea of central origin,” 11 “centrally mediated expiratory apnea with cyanosis,” 12 or briefly “prolonged expiratory apnea” 11 have also been used. The occurrence of PEAC in patients with Chiari II malformation is well-documented in literature. PEAC is a life-threatening condition12–14 that may require tracheotomy and mechanical ventilation.11,15 Additionally, PEAC has been reported in a patient with meningomyelocele who did not have Chiari II malformation. 13 The occurrence of stridor, including inspiratory stridor, has also been documented in patients with Chiari II malformation and meningomyelocele. Respiratory impairment is likely to improve after decompressive surgery. 9 Developmental impairment of the brainstem respiratory network and brainstem compression are possible causes of severe respiratory impairment in patients with Chiari II malformation and meningomyelocele. 16 Accordingly, the arousal response to hypoxia and hypercapnia is critically impaired in patients with Chiari II malformation and meningomyelocele who suffer from apnoea or hypoventilation. 17 Additionally, one study showed that the hypercapnic ventilatory response was reduced in most patients with meningomyelocele. 18 Almost all individuals with a meningomyelocele will have a neurogenic bladder. Cognitive impairment can occur in Chiari II malformation, resulting from hydrocephalus and cerebellar malformation, as the cerebellum is involved in learning and word generation 19 . The prognosis and possible therapies vary depending on the severity of the malformation. However, it has been shown that most patients with neurological impairments benefit from surgical interventions, including the treatment of hydrocephalus with a ventriculoperitoneal shunt, closure of open neural tube defects, and decompression of posterior fossa structures. The timing for the intervention is primarily based on neuroimaging findings and the severity of symptoms.
The treatment of Chiari II malformation requires an interdisciplinary team consisting of “primary care clinicians, neurosurgeons, radiologists, urologists, orthopedic surgeons, and occasionally otolaryngologists” as well as “trained nurses, physical therapists, and pharmacists.” 20 These efforts are significant, as medical problems such as neonatal feeding problems, apnoea, respiratory failure, and neurogenic bowel may persist after surgical intervention. 12
Concerning meningomyelocele, rehabilitative measures vary according to the neurological deficits. Due to in utero degeneration of exposed neural tissue, patients with myelomeningocele often face various neurological deficits depending on the level of the lesion. Foetal surgical repair reduces the occurrence of hydrocephalus and improves neuromotor function compared to postnatal operation. Despite surgical intervention, quality of life during childhood, adolescence, and adulthood are commonly affected. 21 The guidelines of the Spina Bifida Association (SBA) reflect the necessity for an interdisciplinary team to achieve optimal health for patients with spina bifida. They cover the organization of care, psychological and psychosocial issues, neurology, neurosurgery, orthopaedics, urology and sexual health, mobility, physical activity, skin integrity, bowel and bladder management, and sleep disordered breathing. The SBA guidelines also include recommendations to reduce the impact on mental health, to promote family function, and to maximize resilience to spina bifida-related and normative stressors. 22 While most children with spina bifida achieve basic self-care behaviors, they are often 2–5 years behind their typically-developing peers in acquired skills. The SBA guidelines thus also offer recommendations to promote self-management and independence. 23 Furthermore, guidelines on nutrition, metabolic syndromes, and obesity in spina bifida emphasize the importance of good nutrition and physical activity to prevent malnutrition and obesity. 24
Achondroplasia, Chiari II malformation, and meningomyelocele share common ground in management concerning clinical and radiological check-ups to detect compression in the foramen magnum or hydrocephalus, a high level of interdisciplinarity in acute care and rehabilitation alike, and the recommendation of physical activity. 25 The objective of this case report is to describe and discuss a treatment approach that optimized participation in a patient with achondroplasia, Chiari II malformation, and meningomyelocele who suffered from life-threatening respiratory dysregulation. To the authors’ knowledge, the combination of achondroplasia and Chiari II malformation with meningomyelocele has only been reported once in previous literature. 2
Patient information
A three-year-old male suffered from achondroplasia and Chiari II malformation with spina bifida aperta and meningomyelocele. His older sister had been diagnosed with celiac disease; his parents were healthy and aged 33 years when the patient was born. Specifications of his anatomy and neurosurgical procedures are provided under “timeline” and “diagnostic assessment” below. He was referred to the outpatient clinic due to intermittent respiratory arrest in combination with muscular hypertension and loss of consciousness, regularly requiring ventilation with a resuscitation bag and very rarely resulting in admission to a paediatric intensive care unit. These events were triggered by emotions or aspirations, but also occurred suddenly when he played or slept. The patient ate soft food but was coughing after meals. He received non-invasive ventilation (NIV) because of a combination of central apnoea, obstructive apnoea, and nighttime hypercapnia. Oxygen was added to NIV as required between 0.5 l/min. and 5 l/min. Due to a neurogenic bladder with urinary retention and recurrent urinary tract infections, intermittent catheterization twice daily and antibiotic prophylaxis with nitrofurantoin had been implemented. Due to chronic constipation, the patient was treated with macrogol in the evening. The child was attending a daycare facility for children, always in the company of trained assistants who were not nursing professionals. At night, trained nurses at the patient's home provided out-of-hospital intensive care. Speech and language therapy and physiotherapy were performed twice a week, but he did not receive occupational therapy. The patient had an individually adapted set of assistive and therapeutic devices and was mobilized into a standing device daily (Figures 1–4, Table 1).
Patient using his wheelchair.
Patient mobilized into a standing device.

Patient using his walker.
Patient using his therapy chair.
Assistive and therapeutic devices.

Clinical findings
At the first presentation in the outpatient clinic, the child was macrocephalic. He was 69 cm in height and weighed 13 kg. He showed asymmetric dwarfism, had paraplegia at level L3 (American Spinal Injury Association Impairment Scale grade B), and was moving around in the examination room independently in his wheelchair. The patient understood simple commands and communicated with single words. He was anxious during the physical examination, which was disrupted by a sudden respiratory arrest associated with cyanosis. After a short episode of manual ventilation with a resuscitation bag, the patient resumed spontaneous breathing. Coughing was persistently weak.
Timeline
The child was born in October of 2018. In the same month, a ventriculoperitoneal shunt was implanted, and reconstructive surgery of a lumbosacral spina bifida aperta with meningomyelocele was performed. Due to sleep apnoea syndrome, comprising central and obstructive apnoea in combination with nighttime hypercapnia, NIV was initiated in October of 2019. In November of 2020, a spinal decompression and laminectomy were performed because of an occipitoatlantoaxial stenosis.
Diagnostic assessment
A FGFR3-variant (c.1138G > A (p.Gly380Arg)) causing achondroplasia had already been confirmed by Sanger sequencing of exons 3,5,7,8,9,12 and 14 and comparison with the reference sequence.
A cranial MRI from May of 2022 showed a Chiari II malformation with a characteristic small posterior fossa with a low attachment of the tentorium cerebelli. The tectum was typically beaked. Tonsillar herniation into the foramen magnum likely caused the T2 hyperintense lesion of the posterior parts of the medulla oblongata. Additionally, there was a dysgenesis of the corpus callosum (Figure 5). The patient was treated with a left-sided ventriculoperitoneal shunt and sufficient surgical decompression of the foramen magnum. A spinal MRI from May of 2022 showed syringomyelia from C4 downwards (Figure 6) and demonstrated the previous surgical closure of the spina bifida aperta with tethering of the spinal cord to the persistent myelomeningocele (Figure 7).
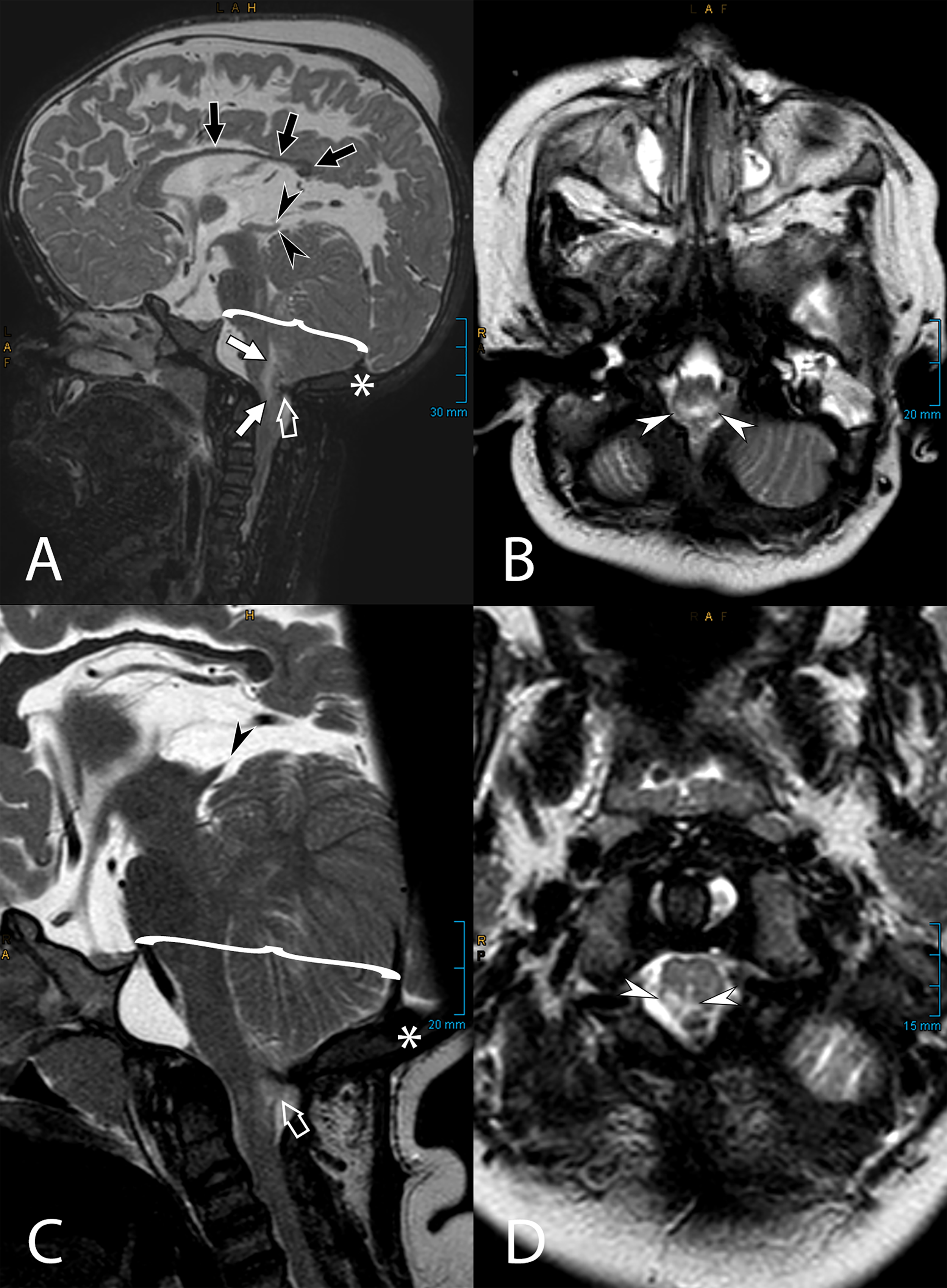
MRI images of a three-year-old patient showing typical findings in Chiari II malformation at the craniocervical junction. All images show a T2-weighted contrast, and orientations are sagittal (A + C) or axial (B + D). A characteristically small posterior fossa is marked by a curved bracket in A + C and is combined with a low attachment of the tentorium cerebelli (white asterisk in A + C). Together with a tethered spinal cord (see Figure 4), this provokes an inferior displacement of the brainstem, leading to a tonsillar herniation into the foramen magnum (open white arrow in A + C). Secondary compression of the medulla oblongata causes a T2 hyperintense lesion in the posterior parts of the medulla, as shown by white arrows (A) and white arrowheads (B + D). A beaked tectum of the midbrain is also typically seen in patients with Chiari II malformation and is marked with black arrowheads (A + C). Additionally, a dysplasia of the corpus callosum is highlighted by black arrows (A).
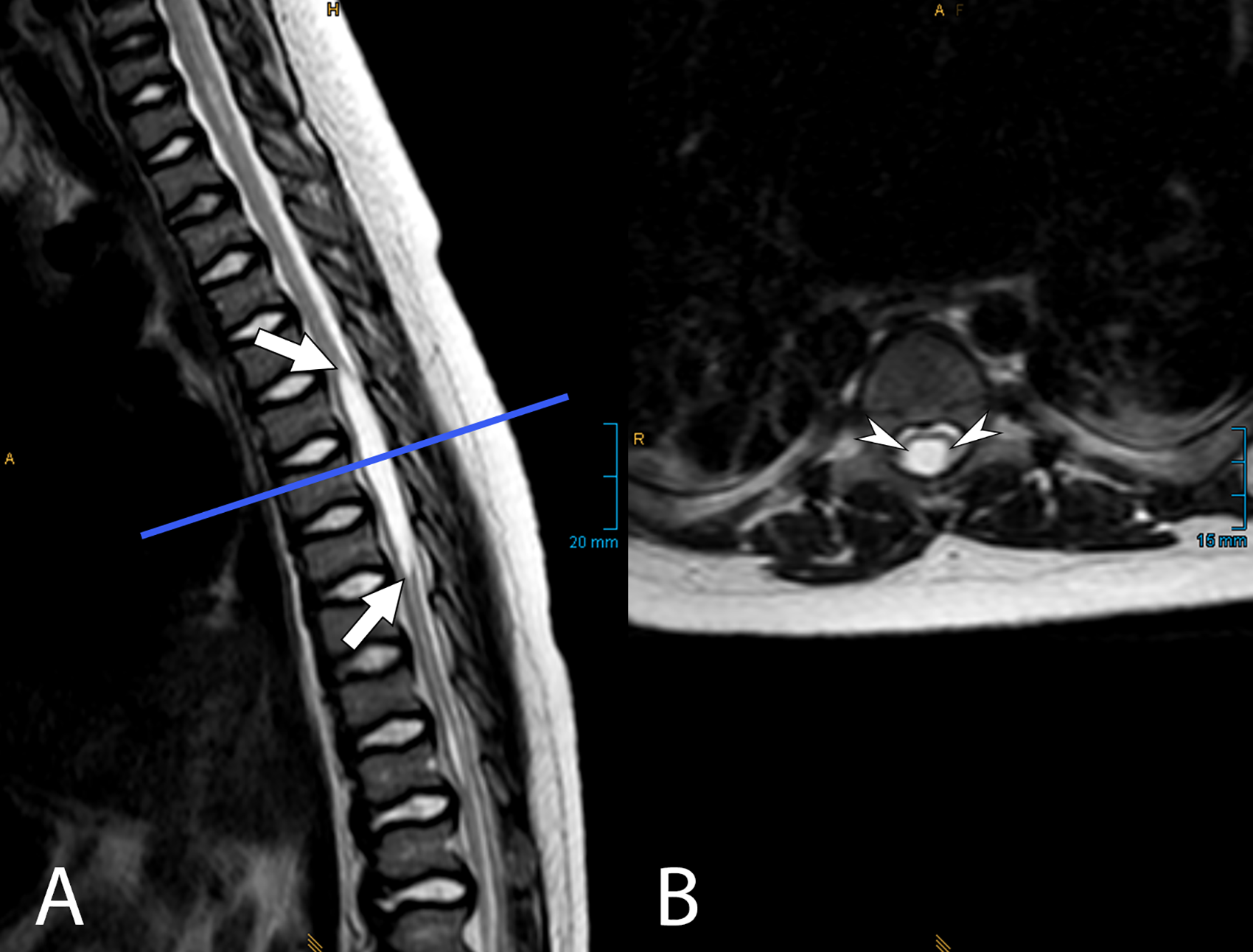
T2-weighted MRI images of the same patient at the thoracic spine level, showing syringomyelia with a fluid-filled cavity at the level of T5-T7 (white arrows in A and white arrowheads in B). The position of the axial slice (B) is indicated by a straight line in the sagittal image (A).
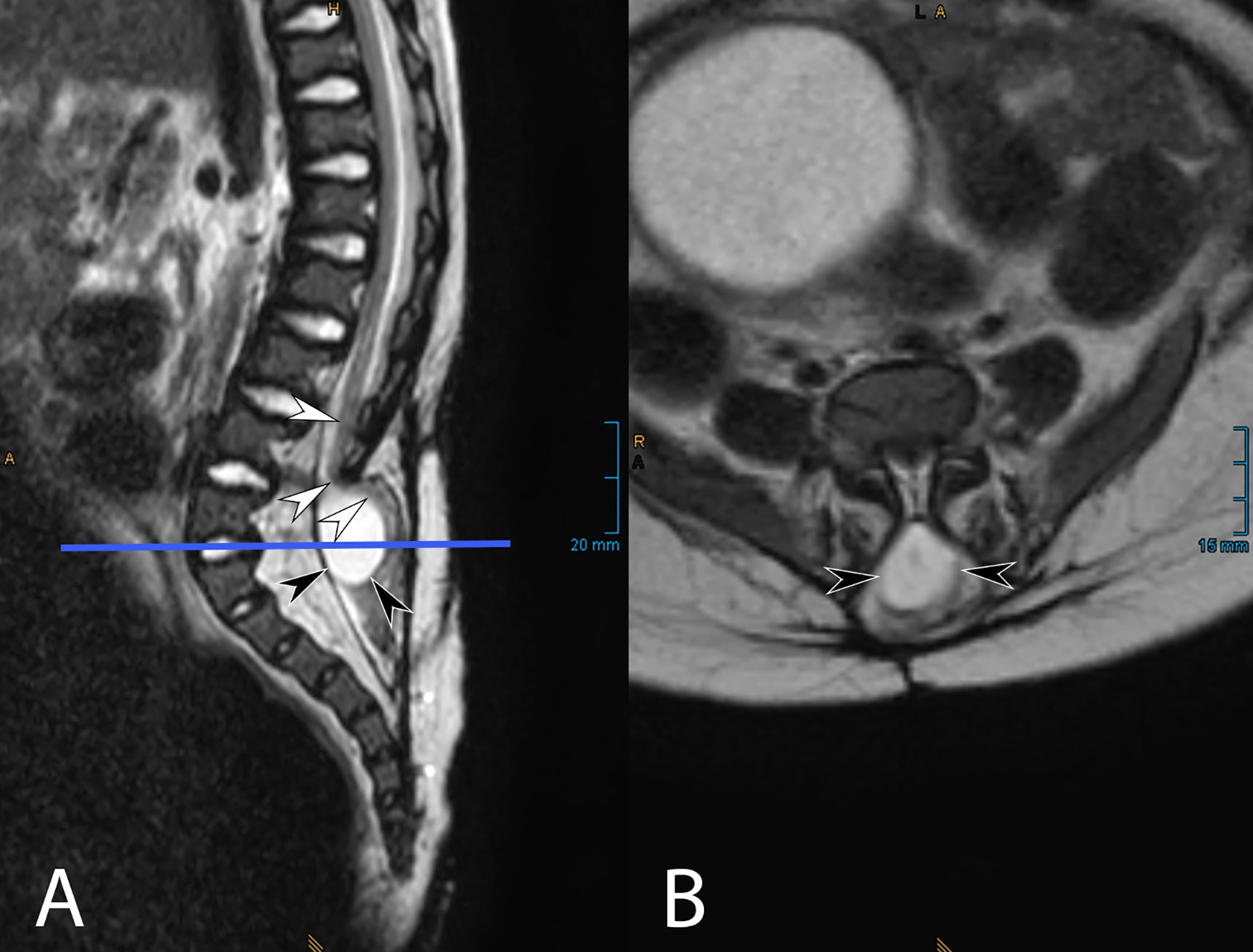
T2-weighted MRI images of the same patient at the lumbar and sacral level, showing the situation after previous surgical closure of the spina bifida aperta with tethering of the spinal cord (white arrowheads in A) to the persistent myelomeningocele (black arrowheads in A + B). The position of the axial slice (B) is indicated by a straight line in the sagittal image (A).
After the parents provided an additional video and sound recording showing predominantly expiratory stridor before and after the event, apnoea in full expiration, and rapid cyanosis, impaired respiratory regulation was suspected and was finally identified as PEAC.
Therapeutic intervention
Oral caffeine (50 mg daily) used as a respiratory stimulant did not reduce the frequency of respiratory events. Tracheostomy was discussed as an ultima ratio but declined, as it would have further compromised language development and limited participation by preventing the patient from attending his daycare facility or, later, kindergarten. Due to a weak cough, mechanical insufflation-exsufflation was started. Regular physiotherapy, speech and language therapy, and mobilization in a standing device were continued, and occupational therapy was initiated. The parents moved to a barrier-free home that was completely maneuvrable with a wheelchair and provided language-driven smart home facilities (See Figures 8 and 9).
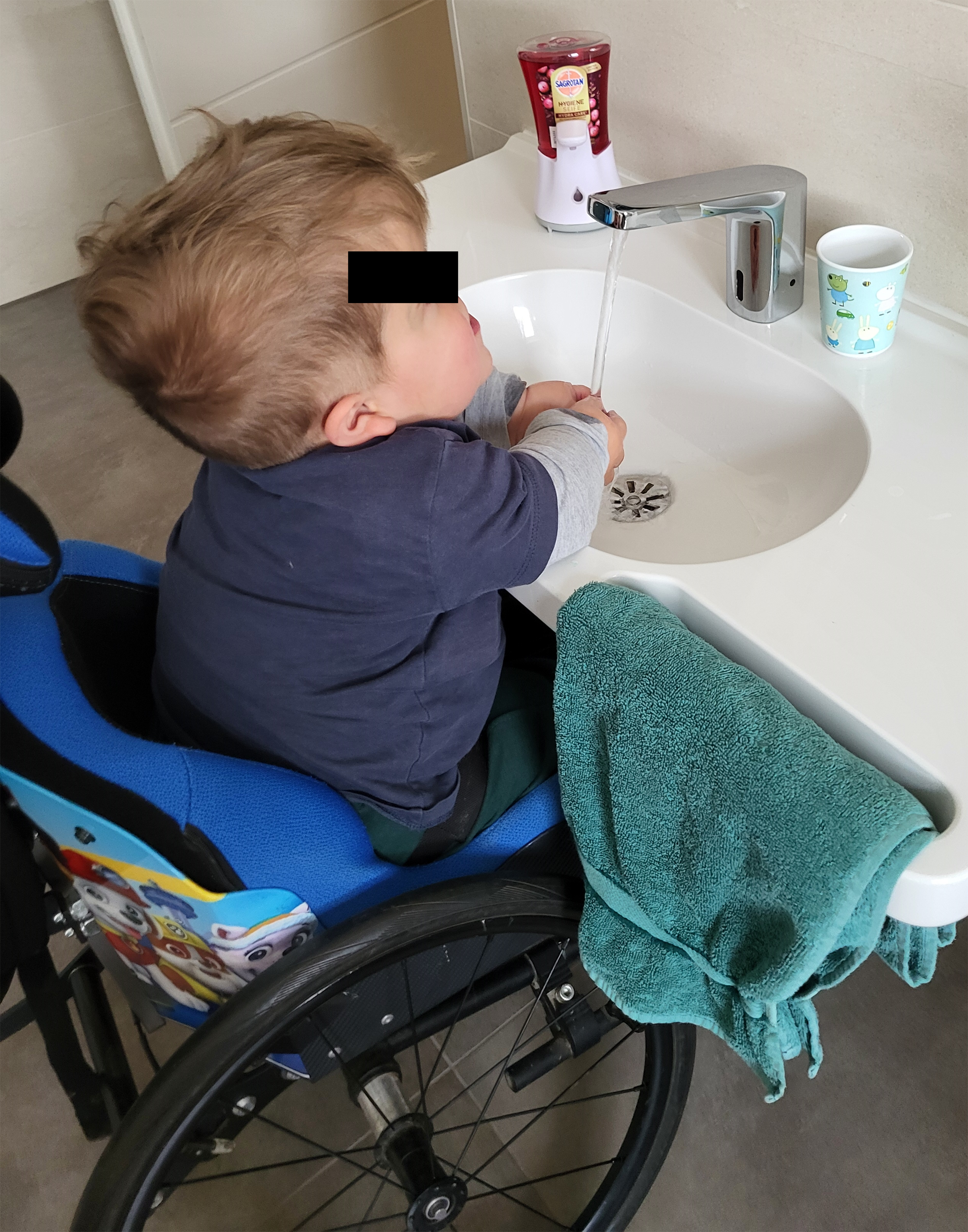
Patient at his individually constructed washbasin.
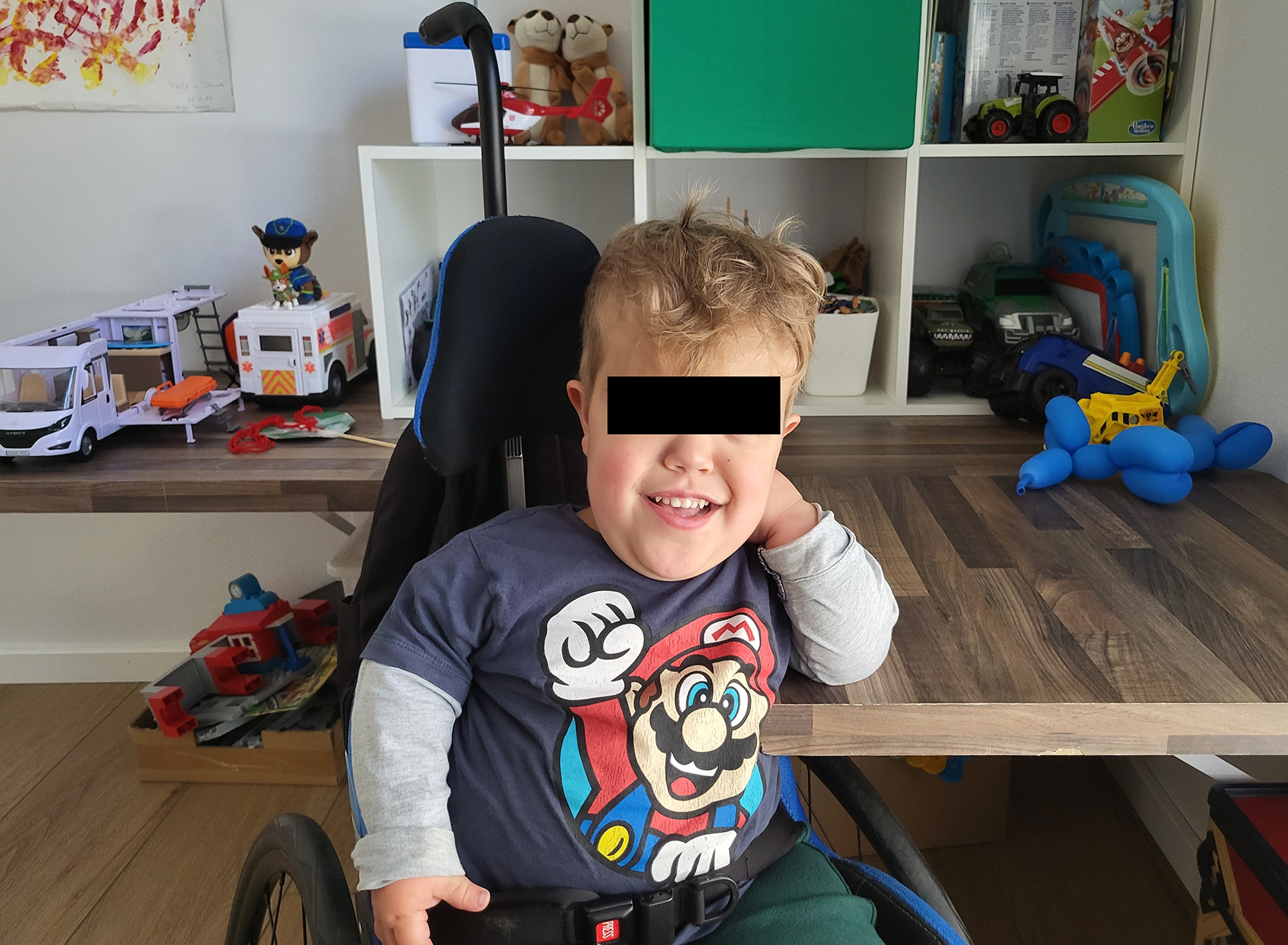
Patient at his roll-under play table.
Follow-up and outcomes
The patient started attending kindergarten just before his fourth birthday. At one-year follow-up, the patient's language capacities had improved, and he could speak four-word sentences. At the two-year follow-up, his parents reported that the last PEAC episode happened five months prior to the follow-up. Language development had improved, and he was able to move around independently with his wheelchair at home, to play in his room without the help of others while still under constant observation, and to perform some activities of daily life like washing his hands independently.
Discussion
The concomitance of achondroplasia and Chiari II malformation is extremely rare. To the authors’ best knowledge, the case presented in this article is the second to have both conditions at the same time, leading to a unique set of health challenges, particularly regarding respiratory management and rehabilitation. While achondroplasia and Chiari II malformation have a variety of symptoms, obesity, hydrocephalus, and respiratory disorders are commonalities. Chiari II malformation causes aspiration, central sleep apnoea, obstructive sleep apnoea, acquired central hypoventilation syndrome, and PEAC, while achondroplasia causes obstructive sleep apnoea and thoracic restrictive disorder. Additionally, achondroplasia and Chiari II malformations can induce similar anatomical changes at the cervicomedullary junction which increase the risk of acute hydrocephalus and brainstem compression. Thus close follow-up to monitor for signs of impeding hydrocephalus are immensely important.1, 2
This patient's MRI from January of 2022 showed a signal hyperintensity at the pontomedullary junction and myelopathy at the height of the foramen magnum. Implantation of a left-sided ventriculoperitoneal shunt and surgical decompression of the foramen magnum had been performed earlier, but still a compression of the brainstem had taken place. In general, it is unclear to what extent brainstem compression or a disorganization of the brainstem respiratory network contribute to respiratory symptoms in patients with Chiari II malformation and meningomyelocele. 16 Notably, respiratory symptoms improve after surgical decompression in most cases, but they may recur. 9
Respiratory impairment was particularly complex in this case. It affected respiratory rhythmogenesis resulting in central sleep apnoea, hypercapnic ventilatory drive resulting in acquired central hypoventilation syndrome, and the respiratory motor pattern resulting in PEAC. Central sleep apnoea might have resulted from a lesion of the ventral respiratory column, particularly the pre-Bötzinger complex, which is the inspiratory pacemaker of the respiratory rhythm in mammals. It is located in the rostral medulla, as is the retrotrapezoid nucleus (RTN), the region best characterized by central chemoreception. RTN regulates CO2 by controlling the breathing frequency and consecutively minute ventilation in slow-wave sleep and quiet wake states. The patient had been diagnosed with hypercapnia at night, possibly resulting from a lesion of the RTN. Lastly, PEAC might result from a lesion of the nucleus retroambiguus, which is in “the medullary ventrolateral tegmentum, rostrocaudally between the obex and the first cervical spinal segment” and has been shown in animal models to coordinate vocal cord motility and abdominal press. 26 Obstructive sleep apnoea, cough insufficiency, and dysphagia further compromised the patient's respiration.
Therefore, nighttime NIV, mechanical insufflation-exsufflation, and continuous supervision by trained personnel were necessary. Tracheostomy is performed as an ultima ratio in patients with Chiari II malformation. However, it was possible to avoid tracheostomy in this case, as the parents were well-trained in applying manual and mechanical ventilation to manage PEAC. The frequency of PEAC episodes decreased during the disease course for this patient, possibly due to regenerative processes in the brain stem and/or neuroplasticity. Respiratory treatments and continuous supervision by trained personnel may not contribute to restorative processes but are important to buy time for those processes to occur. Tracheostomy again would have prevented the patient from attending kindergarten and would have aggravated his language development delay, 27 and later decannulation is rarely possible. 28 Exercise has been shown to improve breathing in patients with achondroplasia, to reduce cardiovascular risk, to avoid obesity, and to improve mental activity and participation in everyday life and overall mental well-being. 4 Accordingly, this patient received occupational therapy, physiotherapy, and mobilization in a standing device, and he was trained using a walker.
In summary, the combination of achondroplasia, Chiari II malformation, and meningomyelocele necessitated an individualized approach to rehabilitation. The combination of life-threatening respiratory impairment, language development delay, paraplegia, neurogenic bladder, and dwarfism required time-consuming, interdisciplinary rehabilitative efforts. These efforts involved various medical specialties, including physiotherapy, speech and language therapy, occupational therapy, respiratory therapy, personal assistance, and out-of-hospital intensive care nursing. Numerous assistive and therapeutic devices were also required, and environmental factors had to be adapted. However, the cornerstones of successful rehabilitation were the parents, who were trained to apply NIV and manual ventilation and could distribute their knowledge to formerly untrained staff. This implies that rehabilitative efforts should be patient-centred and family-oriented to achieve the best possible result. For this patient, the fundamental goals of rehabilitation, namely to reduce the impact of disability and to facilitate equal participation, were widely achieved. The patient could participate in social activities, attend kindergarten, and prepare for elementary school. Complex disability generally requires an ever-adapting, communicative, and eager-to-learn interdisciplinary team to provide optimum care.
Conclusion
The combination of osseous, spinal, and cerebral disease coupled with respiratory impairment presents a complex challenge for paediatric rehabilitation. Interdisciplinary treatment, assistive and rehabilitative technologies, a barrier-free home, and a developmentally appropriate everyday environment are required. The continuous presence of trained caregivers is indispensable when life-threatening events occur or when life-supporting technologies are present. While spinal rehabilitation often incorporates the use of life-supporting technologies, the rehabilitative strategy for patients with a combination of spinal and cerebral pathology, particularly those requiring life-supporting technologies or interventions, remains a topic that requires further understanding and research.
Parents’ perspective
The parents, who played a crucial role in the patient's care, expressed their satisfaction with the progress observed during the 18 months covered by this case report. They noted a significant decrease in the frequency and severity of PEAC episodes, along with an improvement in the patient's cognition. These positive changes strengthened their hope for their child to lead a self-determined life. They also voiced their concerns about needing more nursing professionals in Germany, which compromised inpatient and outpatient care. They believed that it is necessary to reduce administrative and organizational barriers to out-of-hospital intensive care and to foster inclusion through attending kindergarten.
Footnotes
Acknowledgements
No funding was acquired for this article from any sources. The authors would like to thank the patient and his parents for their valuable support.
Ethical considerations
As a case report and literature review, this article is exempt from Institutional Review Board approval. The patient's parents provided informed consent for the publication of the complete manuscript with all figures.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The patient's parents did not give written consent for additional data to be shared publicly, so due to the sensitive nature of the research, supporting data is not available.