
Abstract
Background
Lung cancer remains one of the leading causes of cancer-related mortality worldwide. Holocytochrome c synthase (HCCS), a mitochondrial enzyme involved in apoptosis and energy metabolism, has been implicated in tumorigenesis; however, its role in lung cancer is not well defined.
Aim
This study aimed to elucidate the prognostic and therapeutic potential of HCCS in lung cancer through integrative bioinformatics analyses.
Methods
Transcriptomic, methylation, and clinical data from TCGA were analyzed using TNMplot, UALCAN, and TIMER2.0 to evaluate HCCS expression, promoter methylation, immune infiltration, and prognostic relevance in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC). Meta-analysis of 18 independent cohorts from LUNG CANCER EXPLORER and four GEO datasets validated expression patterns, while Kaplan–Meier analysis assessed survival outcomes.
Results
HCCS was significantly upregulated in LUAD and LUSC and showed promoter hypermethylation in LUAD. Meta-analysis and external validation confirmed its overexpression. Patients with low HCCS expression exhibited poorer overall survival, suggesting a potential tumor-suppressive effect. HCCS expression positively correlated with immune cell infiltration and co-expressed genes enriched in mitochondrial and apoptotic pathways.
Conclusion
HCCS may serve as a prognostic and therapeutic biomarker in lung cancer, linking mitochondrial regulation, epigenetic modification, and immune interactions.
Introduction
Lung cancer remains a leading cause of cancer-related mortality worldwide, accounting for a significant proportion of cancer deaths annually. 1 Despite advancements in diagnostic and therapeutic strategies, the prognosis for lung cancer, particularly in advanced stages, remains poor due to late diagnosis and high metastatic potential. 2 Understanding the molecular mechanisms underlying lung cancer progression is crucial for identifying novel diagnostic biomarkers and therapeutic targets. Although significant progress has been made in cancer therapies in recent years, the overall survival rate for lung adenocarcinoma continues to remain excessively low.3–5 Importantly, immunotherapies—particularly immune checkpoint inhibitors (ICIs)—have been approved as standard first-line and salvage treatment strategies for patients with advanced or recurrent lung cancer.6–9 Nevertheless, most patients with lung cancer exhibit limited responsiveness to ICI therapy, underscoring the urgent need for reliable biomarkers that can enhance diagnosis, guide prognosis, and inform treatment strategies—especially those capable of predicting or modulating immunotherapy outcomes.10–12
Mitochondrial dysfunction is a pivotal factor in cancer biology, influencing processes such as apoptosis, energy metabolism, and reactive oxygen species (ROS) production. 13 Holocytochrome c synthase (HCCS), a nuclear-encoded mitochondrial enzyme, plays a crucial role in cellular bioenergetics and apoptosis regulation by catalyzing the covalent attachment of heme to apocytochrome c, thereby generating holocytochrome c, an essential component of the electron transport chain and the intrinsic apoptotic pathway.14,15 HCCS has attracted significant attention as a key enzyme in the eukaryotic cytochrome c biosynthesis pathway, where it plays an essential role in multiple fundamental biological processes.16–18 Dysregulation of HCCS has been reported in various diseases and cancers, including breast, 19 colorectal, cervical cancer and gastric cancer, where it has been associated with aggressive tumor phenotypes and poor clinical outcomes.20–25 Emerging evidence suggests that aberrant HCCS expression contributes to aggressive tumor behavior and unfavorable clinical outcomes, likely through its central role in mitochondrial cytochrome c maturation, regulation of apoptosis, and cellular energy metabolism.14,15 These biological functions position HCCS as a potential biomarker and mechanistic driver of cancer progression rather than merely a passive indicator of disease status. Aberrant expression or malfunction of HCCS may disrupt mitochondrial respiration and apoptotic signaling, promoting tumor cell survival and progression through evasion of programmed cell death. Moreover, emerging evidence suggests that epigenetic mechanisms, such as promoter DNA methylation, could regulate HCCS expression, contributing to gene silencing frequently observed in cancer, including that of tumor suppressor genes.26–28 However, its specific role in lung cancer, particularly in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), remains inadequately characterized.
High HCCS expression in LUAD correlates with elevated M2 macrophage infiltration and altered immune responses, affecting cancer progression and therapy outcomes. 29 This indicates that HCCS plays a crucial role in modulating the tumor immune microenvironment and represents a potential therapeutic target. 29 Considering the significance of mitochondrial dynamics in tumor biology, HCCS expression may impact cancer cell survival, metabolic reprogramming, and therapy resistance, potentially regulated by DNA methylation. 28 Immune evasion is a hallmark of cancer, and mitochondrial proteins are increasingly recognized as modulators of immune responses within the tumor microenvironment. 30 It is therefore important to investigate whether HCCS expression correlates with immune cell infiltration in lung tumors, which could reveal its involvement in shaping tumor-immune interactions. 29
Epigenetic modifications, particularly DNA methylation, play a critical role in regulating gene expression in lung cancer.28,31 Hypermethylation of promoter regions in tumor suppressor genes often results in their transcriptional silencing, thereby facilitating oncogenesis. With respect to HCCS, promoter methylation has been reported in several other malignancies, indicating that comparable mechanisms may also influence its regulation in lung cancer. Investigating the epigenetic control of HCCS may therefore provide new insights into regulatory pathways that drive tumor initiation, progression, and metastasis.
Recent advances in bioinformatics have revolutionized cancer research by enabling the large-scale integration and interpretation of omics data to identify prognostic and therapeutic biomarkers. Publicly available databases such as The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) provide comprehensive genomic, transcriptomic, and epigenomic datasets that facilitate the systematic exploration of cancer-associated genes across diverse patient cohorts. Through computational pipelines involving differential expression analysis, survival modeling, methylation profiling, and immune infiltration assessment, bioinformatics allows researchers to uncover molecular signatures linked to tumor progression, patient survival, and drug response.32–36 These approaches not only enhance our understanding of cancer biology but also accelerate the discovery of novel diagnostic markers and therapeutic targets, paving the way toward precision oncology and personalized treatment strategies.36–39 In this context, exploring the bioinformatic landscape of HCCS, a mitochondria-associated enzyme implicated in apoptosis and energy metabolism, may provide new insights into its biological function and clinical relevance in lung cancer
The present study aims to comprehensively investigate the expression, methylation, immune infiltration patterns, and gene interaction networks of HCCS in lung cancer computationally, utilizing large-scale transcriptomic, methylation, and clinical datasets from TCGA and GEO. While HCCS dysregulation has been implicated in several malignancies, its role in LUAD and LUSC remains largely unexplored, and no systematic pan-cancer analysis has yet defined its molecular and clinical relevance in these subtypes. By integrating multidimensional data, this study seeks to elucidate the potential tumor suppressive functions of HCCS in LUAD and LUSC, evaluate its prognostic significance, and explore its utility as a therapeutic target. Furthermore, our findings are contextualized within the broader framework of mitochondrial regulators in cancer biology, thereby filling a critical knowledge gap regarding the contribution of HCCS to lung cancer pathogenesis. Our analytical workflow is briefly outlined in Figure 1.
Study flowchart. UALCAN, University of ALabama at Birmingham CANcer data analysis Portal; GTEx, Genotype-Tissue Expression; TIMER, Tumor Immune Estimation Resource; SMART, Shiny Methylation Analysis Resource Tool; GEO, Gene Expression Omnibus.
Material and method
Analysis of HCCS expression across multiple cancer types using TCGA data
To comprehensively assess the expression patterns of the HCCS gene across various tumor types, RNA sequencing data from The Cancer Genome Atlas (TCGA) were systematically analyzed. Initially, the TNMplot platform (https://tnmplot.com/analysis/) was employed to evaluate HCCS expression levels in normal, tumor, and metastatic tissues using its pan-cancer visualization and gene signature analysis tools. This platform provided a comparative overview of HCCS expression dynamics across diverse malignancies, facilitating the identification of potential expression trends associated with tumor progression. 40 Further in-depth analysis was conducted using the TIMER2.0 web tool (http://timer.cistrome.org/), which enables the interrogation of gene expression and immune cell infiltration within the TCGA dataset. Specifically, the “Gene_DE” module was utilized to investigate differential mRNA expression of HCCS between primary tumor tissues and adjacent normal tissues across multiple cancer types. TIMER2.0 integrates standardized RNA-seq data, offering a robust framework for evaluating gene regulation within the tumor microenvironment. This analytical approach allowed for the systematic assessment of HCCS dysregulation in the context of tumor biology and immune interactions. 41 To specifically examine HCCS expression in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), the Gene Set Cancer Analysis (GSCA) platform (https://guolab.wchscu.cn/GSCA/#/expression) was employed. GSCA facilitated the comprehensive analysis of HCCS expression in different clinical stages and molecular subtypes of LUAD and LUSC, providing further insights into the molecular heterogeneity associated with lung cancer progression. GSCA was utilized to assess the expression of ZW10 across varying stages and subtypes of lung cancer, enabling comparative analysis within the same dataset. 42 Statistical significance was evaluated using the Mann–Whitney U test, with p < 0.05 considered statistically significant for all comparisons.
Analysis of HCCS expression and immune infiltration profile in lung cancer
To comprehensively evaluate the expression profile of HCCS in lung cancer, we employed several web-based platforms, including UALCAN, GSCA, GEPIA2, TIMER2.0, and TNMplot, all of which provide access to transcriptomic datasets from TCGA. These resources enabled a broad assessment of HCCS expression across clinical and pathological subgroups of lung adenocarcinoma and lung squamous cell carcinoma. In particular, TIMER2.0 was used to investigate the relationship between HCCS expression and the tumor immune microenvironment. Importantly, the analyses were performed with the “purity adjustment” option enabled, ensuring that correlations between HCCS and immune cell infiltration were not confounded by tumor purity, a major source of bias in immune infiltration studies (LUAD, n = 515 and LUSC, n = 501). Expression levels were visualized using box-and-whisker plots, allowing direct comparisons between tumor and adjacent normal lung tissues. This integrative, purity-adjusted approach highlighted distinct expression patterns of HCCS associated with lung cancer progression, as well as molecular and histological subtypes, underscoring its potential as a clinically relevant biomarker in lung cancer.37,38,43–45 Statistical significance was determined using the Mann–Whitney U test with p < 0.05.
Survival analysis based on HCCS expression in lung cancer
To evaluate the prognostic impact of HCCS expression in lung cancer, survival analyses were conducted using the Kaplan–Meier Plotter (http://www.kmplot.com), a comprehensive online platform that integrates gene expression data with clinical outcomes.44,46 The analysis included data on fast progression (FP) for 1252 patients and overall survival (OS) for 2166 patients, stratified by median HCCS mRNA expression levels. Patients were categorized into high and low expression groups, and Kaplan–Meier survival curves were generated to assess the relationship between HCCS expression and clinical outcomes, including OS and fast progression (FP). Hazard ratios (HR), 95% confidence intervals (CI), and log-rank p-values were calculated to quantify the prognostic significance, with statistical significance defined as p < 0.05. Quality control measures were applied to exclude samples failing quality assessments, ensuring the robustness of the survival analysis. To further validate these findings, TCGA-derived survival data were analyzed using the UALCAN platform (http://ualcan.path.uab.edu), enabling subgroup-specific analysis based on clinical parameters such as menopausal status. This multi-platform approach provided a comprehensive evaluation of the prognostic value of HCCS expression in lung cancer, highlighting its potential as a biomarker for patient stratification and outcome prediction.43,44
Assessment of promoter DNA methylation profile of HCCS in lung cancer
To elucidate potential epigenetic mechanisms underlying the regulation of HCCS expression in lung cancer, promoter DNA methylation levels were systematically analyzed using the UALCAN web platform (http://ualcan.path.uab.edu), which integrates TCGA-derived methylation datasets. This analysis focused on lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), enabling a comparative evaluation of methylation patterns in the HCCS promoter region across these subtypes.
Methylation intensity was quantified using beta values, ranging from 0 (indicative of no methylation) to 1 (indicative of complete methylation), and providing a continuous measure of promoter methylation status. This approach facilitated the identification of epigenetic alterations associated with aberrant HCCS expression, potentially implicating promoter hypermethylation or hypomethylation as regulatory mechanisms in lung cancer pathogenesis. By correlating methylation profiles with transcriptomic data, the study aimed to uncover potential links between promoter methylation and HCCS gene expression dysregulation in lung cancer tissues.38,43
The relationship between DNA methylation levels and gene expression was assessed using the gene-level correlation module of the SMART (Shiny Methylation Analysis Resource Tool) platform (http://www.bioinfo-zs.com/smartapp), an interactive web-based application designed for integrated DNA methylation analysis and visualization based on TCGA datasets. 47 All analyses were performed on October 21, 2025. The dataset included 30 normal and 458 tumor samples for LUAD, and 41 normal and 364 tumor samples for LUSC, all obtained from the TCGA database. For this analysis, the following settings were applied: Analysis method—Mean; Methylation value—M-value; and Correlation coefficient—Pearson.
Gene correlation analyses of HCCS expression in LUAD and LUSC
To identify potential functional partners and co-expressed genes associated with HCCS in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), transcriptomic correlation analyses were conducted using the LinkedOmics platform (https://www.linkedomics.org). This resource enables large-scale, TCGA-based correlation studies by integrating RNA sequencing data across multiple cancer types. Pearson correlation coefficients were calculated to assess the relationship between HCCS expression and all other protein-coding genes within the LUAD and LUSC datasets. Genes exhibiting statistically significant positive or negative correlations with HCCS expression were identified as potential regulatory or functionally related targets. To minimize data variability and enhance analytical robustness, genes with extremely low expression levels (Z score <−4) were excluded from the analysis, ensuring a more reliable interpretation of gene-gene associations. This comprehensive correlation analysis provides insights into the molecular networks potentially modulated by HCCS in lung cancer. 48
Meta-analysis of HCCS expression in lung cancer
We further performed a meta-analysis integrating 18 independent cohorts using lung cancer explorer (https://lce.biohpc.swmed.edu/lungcancer/) to verify the robustness of HCCS expression in lung cancer. 49 The included datasets were derived from multiple transcriptomic platforms, with the majority generated using microarray technology. Quality control of gene expression data was conducted to ensure reproducibility across studies using a method based on the integrative correlation coefficient (ICC).50,51 All datasets underwent standardized preprocessing procedures, including log2 transformation when the data followed a log-normal distribution, followed by quantile normalization at the sample level and global normalization to achieve a mean of zero and unit variance. Gene expression values were subsequently summarized to obtain a single representative expression value per gene for each sample. For each study, the standardized mean difference (SMD) and corresponding 95% confidence interval (CI) were calculated to estimate the effect size of HCCS expression between the two groups. A random-effects model was applied to account for potential heterogeneity among studies. Heterogeneity was assessed using the I2 statistic and Cochran's Q test. A p-value <0.05 was considered statistically significant.
Validation of HCCS expression in independent datasets
To validate the expression pattern of HCCS in lung cancer, we analyzed four independent microarray datasets—GSE19188 (tumor = 91, normal = 65), GSE10072 (tumor = 58, normal = 49), GSE31210 (tumor = 204, normal = 20), and GSE18842 (tumor = 46, normal = 45)—obtained from the Gene Expression Omnibus (GEO) database. Following standard preprocessing and normalization, the expression values of HCCS were extracted from each dataset. Differential expression between tumor and normal samples was assessed using Welch's two-sample t-test, which accounts for unequal variances between groups. Box-and-whisker plots were generated to visualize the results, where the boxes represent the interquartile range (IQR), and the whiskers extend to 1.5 × IQR from the quartiles. The red dot indicates the group mean, with the adjacent number denoting the mean expression value. Statistical significance was defined as p < 0.05 for all comparisons.
Validation of the prognostic value of HCCS in lung cancer
The prognostic significance of HCCS expression in lung cancer was further validated using two independent datasets, GSE3141 and GSE30219, obtained from the Gene Expression Omnibus (GEO) database. Patients were stratified into high and low expression groups based on the median expression level of HCCS within each dataset. Overall survival (OS) analyses were performed using the Kaplan–Meier method implemented in the survminer and survival packages in R. Differences in survival between groups were evaluated using the log-rank test. In addition, Cox proportional hazards regression models were applied to estimate the hazard ratio (HR) and 95% confidence interval (CI) for continuous HCCS expression values in each dataset. A p-value <0.05 was considered statistically significant.
Statistical analysis
All statistical analyses were conducted using a standardized significance threshold, with p-values less than 0.05 considered statistically significant. For survival analyses, the log-rank test was employed to assess differences in survival distributions between groups, with hazard ratios (HR) and corresponding 95% confidence intervals (CI) calculated to quantify the relative risk associated with HCCS expression. The level of statistical significance was denoted using the following conventions: ns (not significant), * (p < 0.05), ** (p < 0.01), and *** (p < 0.001). This systematic approach ensured rigorous assessment of the prognostic impact of HCCS expression in lung cancer, accounting for potential confounding variables and enabling robust interpretation of survival data
Results
Expression profile of HCCS in TCGA cancer types
To investigate the potential involvement of HCCS in tumorigenesis, we systematically assessed its expression across a spectrum of cancer types using TCGA datasets. Comparative analyses between tumor tissues and corresponding adjacent normal samples revealed a notable upregulation of HCCS in several malignancies, underscoring its potential role in cancer progression (Supplementary Figure 1A and B). However, the differential expression patterns varied across tumor types, suggesting a context-dependent regulation of HCCS expression.
To substantiate these findings, pan-cancer analysis was further conducted using the Human Protein Atlas, providing additional validation of HCCS overexpression in multiple malignancies. Elevated HCCS expression was particularly evident in cancers originating from the adrenal gland, bladder, breast, esophagus, liver, lung, ovary, pancreas, prostate, rectum, kidney, skin, stomach, testis, thyroid, and uterus (Figure 2A and B). These observations indicate a potential oncogenic role for HCCS across diverse cancer types and highlight the necessity for further investigation into its mechanistic functions and regulatory pathways in tumorigenesis.
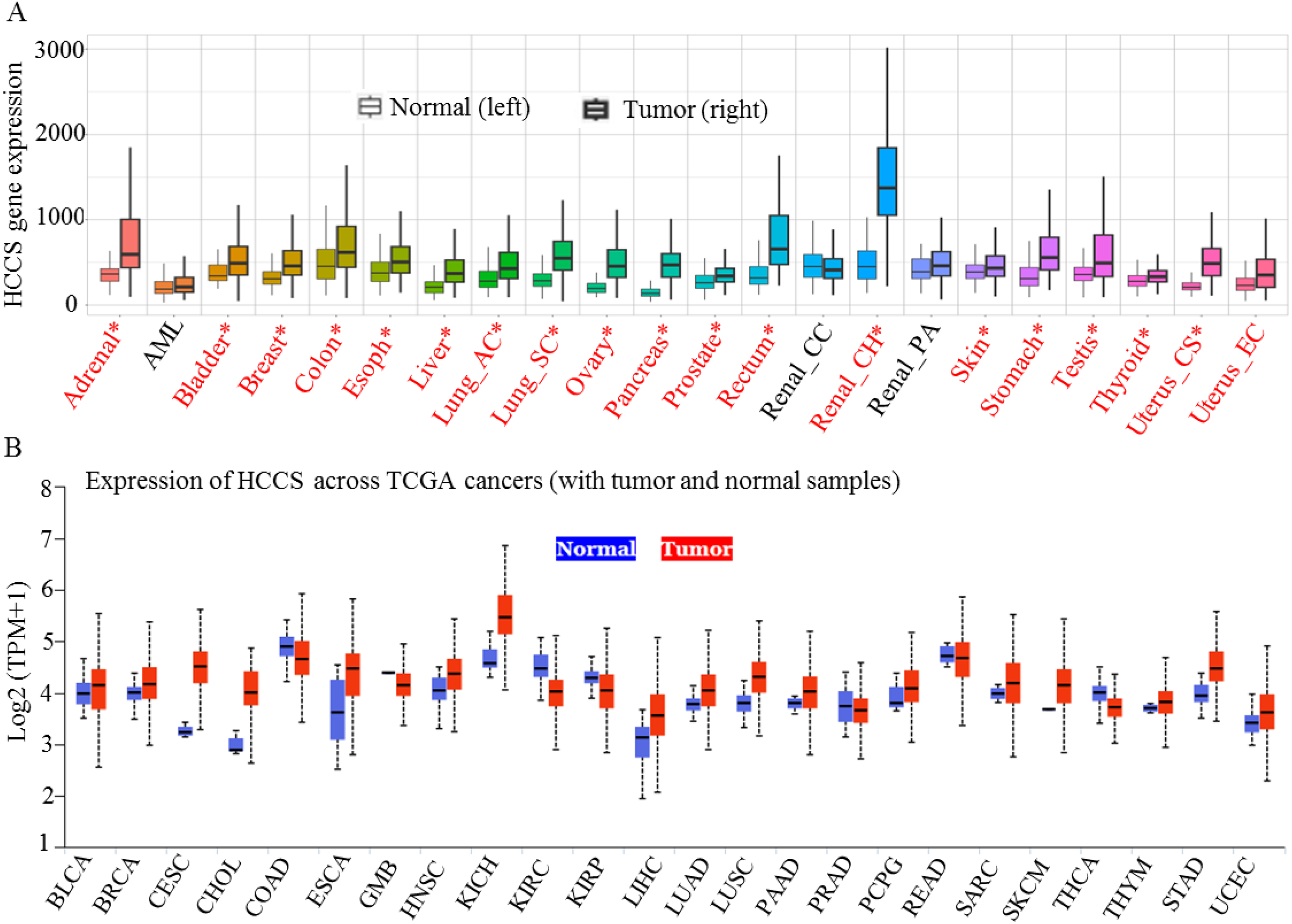
Pan-cancer analysis of HCCS expression using TCGA datasets. (A) The TNMplot tool (https://tnmplot.com/analysis/) was utilized to evaluate the differential expression of HCCS between tumor and normal tissues. Samples with expression values exceeding 10 were included in the analysis. Statistical significance was assessed using the Mann–Whitney U test, with significant differences indicated by red asterisks (*p < 0.05). (B) The UALCAN database (https://ualcan.path.uab.edu/cgi-bin/Pan-cancer.pl?genenam = HCCS) was employed to conduct a comprehensive analysis of HCCS expression across various cancer types. Boxplots illustrate expression levels in tumor tissues (red) compared to normal counterparts (blue), highlighting potential gene dysregulation in different malignancies.
Expression and clinical relevance of HCCS in lung cancer
Following the comprehensive assessment of HCCS expression across multiple cancer types, we further explored its clinical significance within lung cancer, specifically focusing on LUAD and LUSC subtypes. A comparative analysis utilizing TCGA datasets revealed a marked overexpression of HCCS in both LUAD and LUSC tissues relative to adjacent normal tissues (Figure 3A and H). This pronounced upregulation suggests a potential oncogenic role for HCCS in lung cancer and positions it as a promising biomarker candidate for diagnostic and prognostic evaluation.

HCCS expression profiling in lung cancer subtypes. Boxplots depict the expression patterns of HCCS in lung adenocarcinoma (LUAD) (A–G) and lung squamous cell carcinoma (LUSC) (H–N), categorized based on clinical and demographic variables. (A) Comparison of HCCS expression between tumor and normal tissues in LUAD. (B) Analysis of HCCS expression across different nodal metastasis stages in LUAD. (C) HCCS expression stratified by gender in LUAD patients. (D) Influence of smoking status on HCCS expression in LUAD. (E) Expression levels across cancer stages (Stage I–IV) in LUAD. (F) Racial disparities in HCCS expression among LUAD patients. (G) Age-dependent variation in HCCS expression in LUAD. (H) Expression distribution between tumor and normal tissues in LUSC. (I) HCCS expression based on nodal metastasis stages in LUSC. (J) Gender-based expression differences in LUSC. (K) Smoking-related variation in HCCS expression in LUSC. (L) Stage-specific expression analysis (Stage I–IV) in LUSC. (M) Racial stratification of HCCS expression in LUSC. (N) Age-related differences in HCCS expression in LUSC. Statistical significance was assessed using appropriate statistical tests and is denoted as follows: ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001.
To delineate the relationship between HCCS expression and distinct clinical and pathological parameters, we conducted a detailed stratified analysis within LUAD and LUSC subgroups. In LUAD, HCCS expression was examined with respect to nodal metastasis stage (Figure 3B), patient gender (Figure 3C), smoking status (Figure 3D), cancer stage (Figure 3E), race (Figure 3F), patient age (Figure 3G), histological subtypes (Supplementary Figure 2A), and TP53 mutation status (Supplementary Figure 2B). Notably, HCCS expression varied significantly across these clinical parameters, with prominent differences observed across cancer stages and histological subtypes (Supplementary Figures 2C–F).
Similarly, in LUSC, HCCS expression was evaluated across comparable clinical variables, including nodal metastasis stage (Figure 3I), patient gender (Figure 3J), smoking status (Figure 3K), cancer stage (Figure 3L), race (Figure 3M), age (Figure 3N), histological subtypes (Supplementary Figure 2G), and TP53 mutation status (Supplementary Figure 2H). Consistent with LUAD, the expression of HCCS in LUSC demonstrated substantial variation across clinical subgroups, with significant differences observed in tumor stages and histological subtypes (Supplementary Figures 2I–L).
Moreover, further stratified analyses evaluating the association between HCCS expression and additional clinical and pathological parameters confirmed a significant correlation with disease progression in both LUAD and LUSC cohorts (Supplementary Figures 2M, N). These findings underscore the clinical significance of HCCS as a potential biomarker for lung cancer, warranting further investigation to elucidate its mechanistic role in disease progression and its potential as a therapeutic target.
Clinical outcome of HCCS expression in lung cancer
To further investigate the clinical implications of HCCS expression in lung cancer, we performed comprehensive survival analyses using TCGA datasets, focusing on overall survival (OS) and fast disease progression (FP). Kaplan-Meier survival plots were generated using the Kaplan-Meier Plotter platform (www.kmplot.com) with the Affymetrix probe ID: 203745_at, categorizing patients into low- and high-expression groups based on the median HCCS mRNA expression.
The OS analysis demonstrated a significant disparity in survival outcomes between lung cancer patients with high versus low HCCS expression levels. Notably, patients exhibiting lower HCCS expression showed reduced survival rates compared to those with higher expression (n = 2166, HR = 0.71 [0.63–0.8], P = 1.3e-08), underscoring the potential of HCCS as a prognostic marker in lung cancer (Figure 4A). Despite a trend toward increased OS in patients with elevated HCCS expression, the observed differences suggest the need for further investigation in larger cohorts to confirm its prognostic utility.

Prognostic significance of HCCS expression in lung cancer patients. Kaplan–Meier survival analyses were performed to assess the impact of HCCS expression levels on clinical outcomes in lung cancer patients (A and B). (A) Kaplan–Meier curve illustrating overall survival (OS) among 2166 lung cancer patients categorized by HCCS expression levels. (B) Fast progression (FP) analysis based on HCCS expression in 1252 lung cancer patients. Patients were stratified into high and low expression groups using the Affymetrix probe ID 203745_at. Red lines represent the high-expression group, while black lines denote the low-expression cohort. Data were obtained using the Kaplan–Meier Plotter platform (www.kmplot.com). (C–F) Subgroup analyses investigating the relationship between HCCS expression and survival outcomes based on clinical and demographic factors using TCGA data accessed via the UALCAN portal (http://ualcan.path.uab.edu/). (C) Survival analysis in LUAD patients stratified by HCCS expression levels. (D) Influence of smoking status on survival in LUAD patients with varying HCCS expression. (E) Survival analysis in LUSC patients categorized by HCCS expression levels. (F) Effect of smoking status on survival in LUSC patients with differing HCCS expression. Statistical significance was evaluated using log-rank tests, with the following annotations: ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001. “Exp.” denotes expression levels, and “med.” indicates medium expression.
In the context of FP, a significant association was observed between HCCS expression and disease progression. Patients with lower HCCS expression experienced markedly shorter FP relative to those with higher expression levels (n = 1252, HR = 0.67 [0.57–0.8], P = 4.5e-06) (Figure 4B). These findings suggest that HCCS may serves as a potential prognostic indicator for more aggressive disease phenotypes in lung cancer.
To refine our analysis, we further explored survival outcomes in clinical subgroups, including race, gender, and cancer subtype (LUAD and LUSC) (Figure 4C). In LUAD, the survival analysis revealed a notable reduction in survival probability among reformed smokers with low/medium HCCS expression compared to other subgroups (Figure 4D). Additionally, stratified analyses by gender indicated that elevated HCCS expression in female patients was associated with reduced OS compared to males (Supplementary Figure S3A). Racial differences in survival outcomes also emerged, with lower survival rates observed in Caucasian patients with low/medium HCCS expression (Supplementary Figure S3B).
Similarly, in LUSC, patients with high HCCS expression who were smokers exhibited a significantly reduced survival probability compared to non-smokers (P = 0.02) (Figure 4E and F). Gender-specific analysis revealed a similar trend, with female patients displaying poorer survival outcomes relative to males (Supplementary Figure S3C). Furthermore, Asian patients with high HCCS expression showed reduced survival compared to other racial groups (Supplementary Figure S3D).
Collectively, these findings highlight the prognostic significance of HCCS expression in lung cancer, particularly in relation to disease progression and overall survival. The observed associations with clinical subgroups, including smoking status, gender, and race, suggest that HCCS may serves as a valuable stratification biomarker for patient risk assessment. Further studies are warranted to elucidate the underlying molecular mechanisms by which HCCS influences survival outcomes and to explore its potential as a therapeutic target.
Correlation of HCCS expression with immune infiltration profile in lung cancer
To further elucidate the functional implications of HCCS in the tumor microenvironment of lung cancer, we investigated its association with immune cell infiltration using Spearman's correlation analysis. The analysis was conducted using TCGA transcriptomic data to assess the relationship between HCCS expression and the infiltration levels of various immune cell populations in LUAD and LUSC.
Our findings revealed significant positive correlations between HCCS expression and multiple immune cell types, including CD8+ T cells, neutrophils, CD4+ T cells, and monocytes in LUAD (Figure 5A–D) and LUSC (Figure 5E–H). These associations suggest that HCCS may play a role in modulating both pro-inflammatory and immunosuppressive cell populations within the lung cancer microenvironment, potentially influencing immune surveillance and tumor progression.

Association between HCCS expression and immune cell infiltration in lung cancer. Scatter plots depict the Spearman correlation between HCCS expression and the infiltration levels of various immune cell types in LUAD (n = 515) (A–D) and LUSC (n = 501) (E–H), as analyzed using TCGA transcriptomic data. For LUAD: (A) CD8+ T cells, (B) Neutrophils, (C) CD4+ T cells, (D) Monocytes. For LUSC: (E) CD8+ T cells, (F) Neutrophils, (G) CD4+ T cells, (H) Monocytes. Each plot shows a significant positive correlation between HCCS expression and immune cell abundance. The strength and statistical significance of these associations were assessed using Spearman's rank correlation test, with p-values <0.05 considered statistically significant.
In addition, further analyses of HCCS expression in relation to other immune cell populations, such as macrophages, cancer-associated fibroblasts, mast cells, and eosinophils, revealed consistent positive correlations in both LUAD and LUSC (Supplementary Figure S4). These observations underscore the potential involvement of HCCS in shaping the immune landscape in lung cancer and its possible contribution to the tumor-promoting or immunosuppressive phenotypes.
Given the observed upregulation of HCCS in lung cancer, our data suggest that its expression may not only be linked to tumor cell proliferation but also to the regulation of immune cell dynamics in the tumor microenvironment. Future studies are warranted to validate these findings and to investigate the mechanistic basis by which HCCS may influence immune cell recruitment and functional polarization in lung cancer.
Promoter DNA methylation of HCCS in lung cancer and its clinical associations
Epigenetic alterations, particularly promoter DNA methylation, play a critical role in regulating gene expression in cancer. To explore the potential epigenetic regulation of HCCS in lung cancer, we assessed the methylation status of its promoter region across diverse clinical and pathological characteristics using TCGA lung cancer datasets. Beta values, ranging from 0 (unmethylated) to 1 (fully methylated), were employed to quantify methylation levels.
Our analysis revealed significant hypermethylation of the HCCS promoter region in LUAD tissues compared to normal lung tissues (Figure 6A), suggesting a potential regulatory mechanism by which HCCS expression may be modulated in lung tumorigenesis. Notably, HCCS promoter methylation levels varied across several clinical parameters, including nodal metastasis stage, gender, race, cancer stage, smoking status, age, and TP53 mutation status (Figure 6, Supplementary Figure S5). Specifically, HCCS methylation levels were markedly elevated in primary tumors relative to normal lung tissues (p < 0.05) (Figure 6A). Additionally, a progressive increase in methylation was observed in advanced nodal metastasis stages, particularly in N1, N2, and N3 (Figure 6B). Gender- and race-based differences were also detected, with elevated methylation levels in females and among Caucasian, African-American, and Asian patients (Figure 5C, 5D).
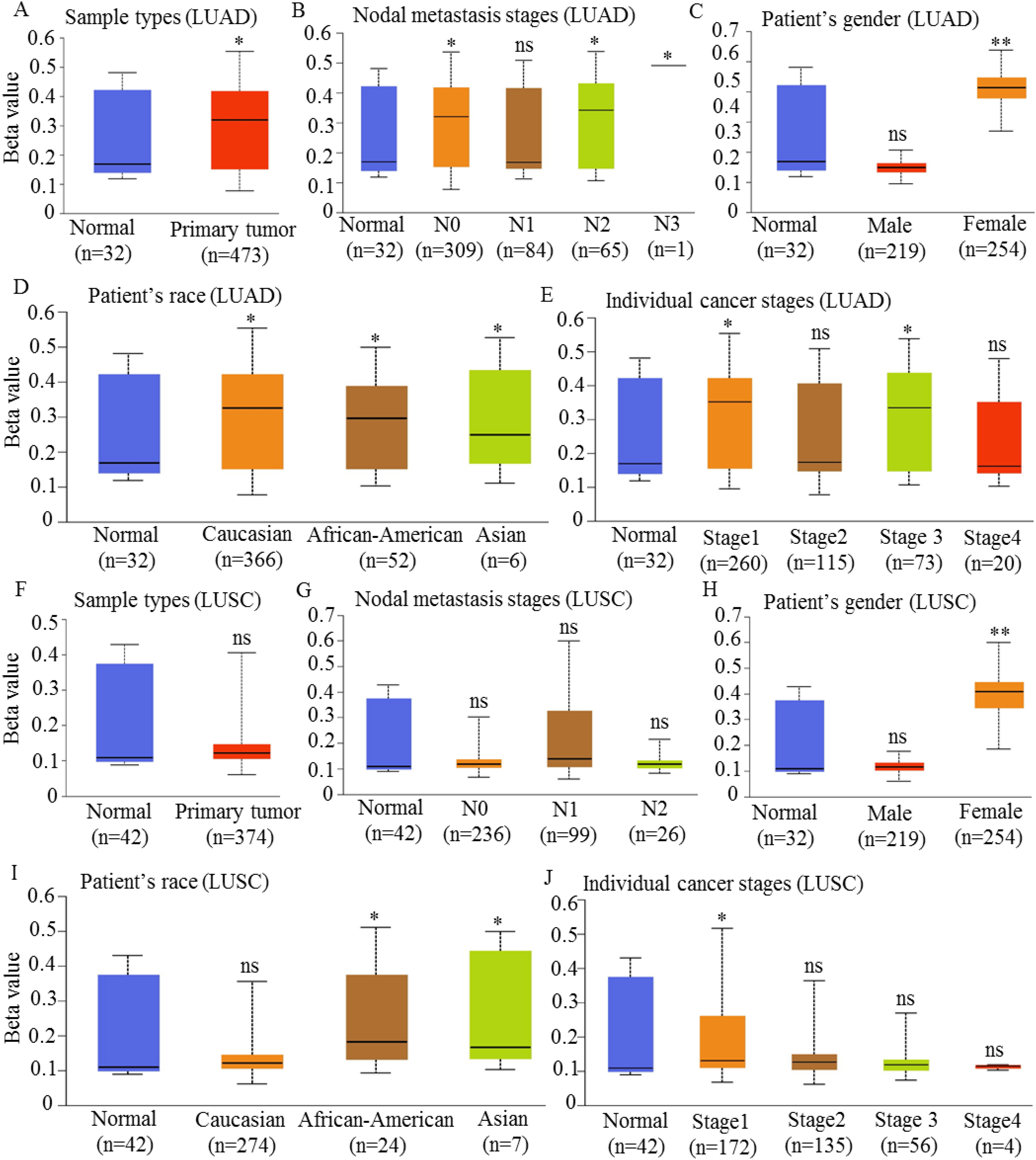
Promoter DNA methylation profile of HCCS in lung cancer subtypes. Boxplots illustrate the promoter DNA methylation levels of the HCCS gene in LUAD and LUSC samples, categorized by clinical and demographic variables. For LUAD: (A) Methylation levels in tumor versus normal tissues. (B) Methylation distribution across nodal metastasis stages. (C) Gender-specific differences in promoter methylation. (D) Racial disparities in promoter methylation levels. (E) Methylation variation across cancer stages (Stage I–IV). For LUSC: (F) Methylation levels in tumor versus normal tissues. (G) Methylation distribution across nodal metastasis stages. (H) Gender-specific differences in promoter methylation. (I) Racial disparities in promoter methylation levels. (J) Methylation variation across cancer stages (Stage I–IV). Methylation data were retrieved from the UALCAN platform, and statistical analyses were performed using appropriate tests. Statistical significance is denoted as follows: ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001.
Further stratified analyses revealed a significant association between HCCS promoter methylation and cancer stage (Figure 6E), smoking status (Supplementary Figure S5A), age (Supplementary Figure S5B), and TP53 mutation status (Supplementary Figure S5C). Of note, TP53 mutations were associated with significantly higher HCCS methylation, suggesting a potential epigenetic-gene interaction in lung cancer pathogenesis.
In contrast, the analysis of HCCS promoter methylation in LUSC revealed no statistically significant differences when compared to normal tissues (Figure 6F). Although higher methylation levels were observed in primary LUSC tumors, the differences were not significant (p > 0.05). Methylation levels across nodal metastasis stages remained relatively stable (Figure 6G), with similar patterns observed in gender and racial subgroups (Figure 6H, 6I). Additionally, no significant associations were observed between HCCS methylation and other clinical parameters, including cancer stage (Figure 6J), smoking status (Supplementary Figure S5D), age (Supplementary Figure S5E), and TP53 mutation status (Supplementary Figure S5F).
Collectively, these findings indicate that promoter hypermethylation of HCCS is observed in LUAD but not in LUSC, highlighting the epigenetic heterogeneity between these lung cancer subtypes. Although promoter hypermethylation is generally associated with gene silencing, our data revealed a concurrent upregulation of HCCS in LUAD. Typically, promoter methylation suppresses gene transcription, whereas gene body methylation is often associated with increased expression. CpG islands located within promoter regions are key regulatory elements that modulate gene activity through methylation-dependent transcriptional silencing. To examine whether promoter methylation influences HCCS expression in lung cancer, we performed a correlation analysis between methylation levels at CpG islands and HCCS mRNA expression using TCGA datasets.
In LUAD, a clear negative trend was observed between promoter methylation and HCCS expression (Supplementary Figure S6A). Eleven CpG sites exhibited negative correlations, while three sites showed positive correlations with gene expression. Overall, HCCS expression demonstrated a weak inverse relationship with promoter methylation (R = –0.066, p = 0.15), suggesting that higher methylation may contribute to modest transcriptional repression.
Similarly, in lung squamous cell carcinoma (LUSC), most CpG probes displayed a negative association between methylation and gene expression (11 negative and 3 positive correlations; Supplementary Figure S6B). The aggregated analysis revealed a weak negative correlation (R = –0.059, p = 0.25), indicating a comparable regulatory pattern to that observed in LUAD.
Although the correlation coefficients were not statistically significant, the overall negative association between promoter methylation and HCCS expression in both LUAD and LUSC supports the notion that DNA methylation may partially suppress HCCS transcription in lung cancer. This finding aligns with the canonical role of promoter hypermethylation in transcriptional downregulation. The modest correlation coefficients, however, imply that additional regulatory mechanisms—such as post-transcriptional or mitochondrial feedback control—may also influence HCCS expression in these tumor types.
Construction and analysis of gene interaction networks for HCCS in LUAD and LUSC
To further elucidate the molecular role of HCCS in lung cancer, we conducted a comprehensive analysis of its gene interaction networks, focusing on LUAD and LUSC subtypes. Given that genes function within intricate networks that regulate critical cellular processes such as cell cycle progression, mitotic control, and tumorigenesis, understanding these interactions can provide valuable insights into the functional significance of HCCS in cancer biology.
We employed gene correlation analyses to identify genes co-expressed with HCCS across lung cancer tissues. Our findings revealed a set of genes positively correlated with HCCS expression in both LUAD and LUSC, suggesting its involvement in regulatory pathways that promote oncogenic signaling, mitotic stability, and metastasis (Figure 7A, 7B). These co-expressed genes may function in concert with HCCS to mediate key oncogenic processes, further underscoring the potential oncogenic role of HCCS observed in previous sections. Conversely, we also identified genes negatively correlated with HCCS expression, highlighting potential regulatory mechanisms that counteract HCCS-mediated oncogenic effects or that function in distinct cellular pathways (Supplementary Figure S7A, S7B). This dichotomy in gene correlations emphasizes the complex regulatory networks involving HCCS and underscores its multifaceted role in lung cancer biology.

Gene expression correlation analysis of HCCS in lung cancer. (A) Expression profile of HCCS in lung adenocarcinoma (LUAD), accompanied by a heatmap displaying the top genes exhibiting a positive correlation with HCCS expression in LUAD samples. (B) Heatmap illustrating the top genes positively correlated with HCCS expression in lung squamous cell carcinoma (LUSC). Correlation analyses were conducted using TCGA LUAD and LUSC transcriptomic datasets, with Pearson correlation coefficients applied to identify statistically significant associations (p < 0.05).
Given the previously noted upregulation of HCCS in multiple cancers, including lung cancer, constructing these gene interaction networks not only advances our understanding of its oncogenic potential but also highlights potential therapeutic targets. Future investigations incorporating pathway enrichment analyses and functional validation assays are essential to substantiate these gene interactions and to elucidate the precise molecular mechanisms through which HCCS modulates tumorigenesis in lung cancer.
Meta-analysis of HCCS expression in lung cancer
To comprehensively evaluate HCCS expression across multiple datasets, we performed a meta-analysis comparing tumor and normal lung tissues using a random-effects model. As shown in Figure 8, most datasets exhibited positive standardized mean differences (SMDs), indicating higher HCCS expression in tumor tissues. The pooled analysis revealed a significant overall upregulation of HCCS in lung cancer (SMD = 0.55, 95% CI = 0.38–0.72, p = 1.0 × 10−10). Moderate heterogeneity was observed among studies (I2 = 63%), suggesting some variability in effect sizes, possibly due to differences in sample size, platforms, or tumor subtypes. Collectively, these findings demonstrate that HCCS is consistently overexpressed in lung cancer compared to normal tissues, consistent with our primary analysis.
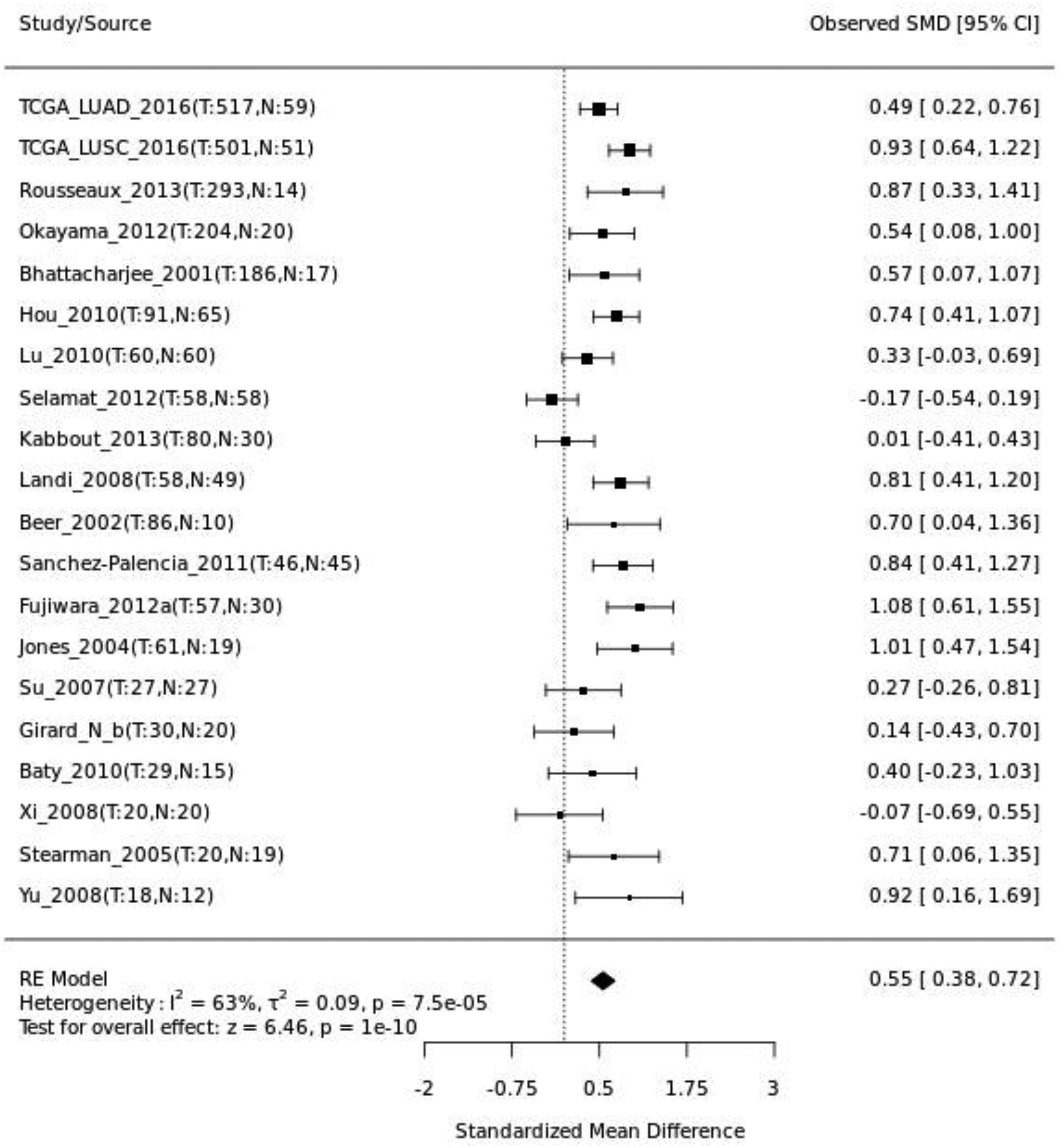
Meta-analysis of HCCS expression in independent cohorts. Forest plot showing the standardized mean differences (SMDs) and 95% confidence intervals (CIs) of HCCS expression between lung tumor and normal tissues across independent datasets. The pooled estimate, calculated using a random-effects model, indicates significantly higher HCCS expression in tumor tissues (SMD = 0.55, 95% CI = 0.38–0.72, p = 1.0 × 10−10), with moderate heterogeneity (I2 = 63%).
Validation of HCCS expression in independent datasets
To further validate the differential expression of HCCS in lung cancer, four independent GEO datasets were analyzed, including GSE19188, GSE10072, GSE31210, and GSE18842. Consistent with our previous findings, HCCS expression was significantly upregulated in tumor tissues compared to normal lung tissues across all datasets (Figure 9A–D). Specifically, GSE19188 showed markedly higher HCCS expression in tumors (p = 1.2 × 10−6), while similar trends were observed in GSE10072 (p = 3.2 × 10−5), GSE31210 (p = 2.4 × 10−5), and GSE18842 (p = 1.4 × 10−4). These consistent results across multiple independent cohorts strongly support that HCCS is significantly overexpressed in lung cancer tissues, suggesting its potential oncogenic relevance in tumor development.

Independent validation of HCCS expression and prognostic significance. A–D: Box plots from four independent GEO datasets (GSE19188, GSE10072, GSE31210, and GSE18842) showing significantly higher HCCS mRNA expression in tumor tissue compared to normal tissue (p-values provided). E and F: Kaplan-Meier overall survival (OS) curves from two independent GEO datasets (GSE3141 and GSE30219) demonstrating that low HCCS expression is significantly associated with poorer overall survival (HR and log-rank p-values provided).
Validation of the prognostic value of HCCS in lung cancer
The prognostic significance of HCCS expression was validated in two independent lung cancer cohorts from the GEO database. Kaplan-Meier survival analysis demonstrated that high expression of HCCS was associated with significantly improved overall survival in both validation datasets (Figure 9E and F). In the GSE3141 cohort, patients with high HCCS expression had a significantly longer overall survival compared to those with low expression (Hazard Ratio [HR] = 0.57, 95% Confidence Interval [CI] 0.34–0.98; log-rank P = 0.039). This finding was corroborated in the GSE30219 cohort, where high HCCS expression similarly conferred a significant survival advantage (HR = 0.75, 95% CI 0.57–0.99; log-rank P = 0.042). Consistent with the Kaplan-Meier results, univariate Cox proportional hazards regression analysis confirmed that increased HCCS expression, treated as a continuous variable, was significantly associated with a reduced risk of death in both datasets. In conclusion, these independent validations consistently indicate that high expression of HCCS is a favorable prognostic factor in lung cancer.
Discussion
This study provides a comprehensive bioinformatics characterization of holocytochrome c synthase (HCCS) in lung cancer by integrating large-scale transcriptomic, epigenetic, and clinical datasets. Consistent across multiple analytic platforms, HCCS was markedly upregulated in both LUAD and LUSC tissues relative to normal controls. To illustrate the integrative interpretation of these results, a mechanistic model (Figure 10) was developed to depict the potential role of HCCS in lung tumorigenesis. The model summarizes how HCCS overexpression, modulated by promoter methylation and associated with immune infiltration and oncogenic signaling pathways, may contribute to tumor progression and clinical outcomes in LUAD and LUSC (Figure 10). To ensure the robustness and reproducibility of our findings, we complemented TCGA-based analyses with extensive independent validation across multiple datasets and platforms. A random-effects meta-analysis of 18 independent cohorts confirmed consistent HCCS overexpression in lung cancer, while four GEO expression datasets independently validated this trend. Furthermore, two external GEO survival cohorts substantiated the prognostic association of HCCS expression with overall survival. This multi-cohort validation framework reinforces that our findings extend beyond a single database and represent a comprehensive, reproducible, and biologically consistent characterization of HCCS in lung cancer.

Mechanistic model of HCCS function in lung cancer. HCCS is upregulated across multiple cancers and notably in LUAD and LUSC, where its expression correlates with clinical stage, TP53 mutation, and patient demographics. Elevated HCCS influences both tumor-intrinsic pathways (cell cycle, mitosis, and metastasis) and the tumor microenvironment through immune infiltration and epigenetic alterations. Promoter hypermethylation in LUAD suggests an altered regulatory mechanism, while stable methylation in LUSC reflects subtype-specific differences. Together, these effects contribute to tumor progression, poor survival, and immune modulation, establishing HCCS as a potential prognostic biomarker and therapeutic target.
Meta-analysis of 18 independent cohorts from the lung cancer explorer platform further substantiated this observation, revealing a significant pooled standardized mean difference (SMD = 0.55, 95% CI = 0.38–0.72, p = 1.0 × 10−10). The consistency of overexpression across heterogeneous datasets underscores the robustness of HCCS dysregulation in lung cancer and supports its biological and clinical relevance. The moderate heterogeneity (I2 = 63%) observed likely reflects variation in experimental platforms, tissue composition, and subtype distribution—common challenges in cross-cohort gene expression meta-analyses. Nevertheless, the random-effects model confirmed the reproducibility of HCCS upregulation, validating its potential as a tumor-associated gene.
Independent validation using four GEO datasets (GSE19188, GSE10072, GSE31210, and GSE18842) confirmed significantly elevated HCCS expression in tumor tissues, thereby strengthening the generalizability of our findings beyond TCGA. This multi-cohort confirmation minimizes dataset-specific bias and enhances translational reliability, as independent datasets often vary in geographic origin, sequencing platforms, and patient demographics. 49 Together, the meta-analysis and validation data collectively provide high-confidence evidence that HCCS is consistently dysregulated in lung cancer.
The prognostic significance of HCCS expression was further verified in two GEO cohorts (GSE3141 and GSE30219), where higher expression levels correlated with significantly improved overall survival. This finding mirrors TCGA survival data, in which patients with low HCCS expression experienced poorer prognosis. This apparent paradox—HCCS being overexpressed in tumors but associated with favorable outcomes—suggests a context-dependent or compensatory role. Elevated HCCS may represent a cellular response to mitochondrial stress or oxidative imbalance, acting protectively to maintain mitochondrial function and promote apoptosis under tumor stress. Similar dual behavior has been observed in other mitochondrial regulators such as LACTB and MPC1, which display tumor-suppressive properties despite overexpression under certain stress conditions.52–54 These results indicate that loss or downregulation of HCCS may impair mitochondrial integrity, promoting tumor survival and aggressiveness.
The observed promoter hypermethylation of HCCS in LUAD, but not in LUSC, indicates subtype-specific epigenetic regulation. This pattern may reflect compensatory gene activation, allele-specific methylation, or partial methylation heterogeneity across CpG islands. Notably, the association between HCCS hypermethylation and TP53 mutation implies coordinated disruption of mitochondrial and nuclear genome stability pathways.55–57 Although promoter hypermethylation typically silences gene expression, certain genes—especially mitochondrial enzymes—can exhibit paradoxical overexpression due to transcriptional feedback loops or demethylation at alternative promoter sites.58,59 To examine its functional effect, we performed a correlation analysis between promoter methylation and HCCS expression using TCGA data via the SMART platform. The results revealed a weak but consistent negative correlation in both LUAD and LUSC, indicating that increased methylation tends to repress HCCS expression. However, despite this inverse trend, HCCS remained upregulated in LUAD, implying that promoter methylation alone does not fully account for its transcriptional activation. This paradox has been observed in other mitochondrial genes and may reflect partial methylation, allele-specific regulation, or compensatory transcriptional activation under metabolic stress. Collectively, these findings suggest that HCCS expression is modulated by both epigenetic and mitochondrial regulatory mechanisms, rather than solely by promoter methylation.
Our immune infiltration analysis revealed that HCCS expression correlates positively with several immune cell types, including CD8+ and CD4+ T cells, macrophages, and mast cells, suggesting that HCCS may modulate the tumor microenvironment. Elevated HCCS expression in LUAD was associated with enhanced infiltration of anti-tumor immune populations, whereas its correlation with macrophages in LUSC may indicate an immunosuppressive role. These findings reinforce the hypothesis that mitochondrial proteins regulate immune activity within tumors.60–62 By influencing immune metabolism, antigen presentation, and cytokine signaling, HCCS may shape both pro- and anti-tumor immune responses, a mechanism increasingly recognized as critical for immunotherapy responsiveness.61,63
Gene co-expression analysis identified HCCS-associated genes enriched in mitochondrial, oxidative phosphorylation, and apoptotic signaling pathways—core regulators of cellular metabolism and tumorigenesis.26,29,54 These networks highlight the central role of HCCS in maintaining mitochondrial homeostasis and its potential interaction with key metabolic pathways that influence both tumor growth and immune response. Together, these findings suggest that HCCS serves as an integrative node linking mitochondrial metabolism, epigenetic regulation, and immune modulation in lung cancer.
Clinical implications
The integrative findings of this study suggest that HCCS could serve as a valuable prognostic and predictive biomarker in lung cancer. Its consistent upregulation, association with better survival, and immune relevance position it as a complementary marker alongside existing clinical parameters such as PD-L1, EGFR, KRAS, and ALK status. Since mitochondrial metabolism has been increasingly linked to immunotherapy outcomes, assessing HCCS expression could help stratify patients likely to benefit from immune checkpoint inhibitors.6–9,60,63 Moreover, given its mitochondrial localization and role in cytochrome c biogenesis, HCCS may represent a novel therapeutic target for restoring apoptotic sensitivity in resistant lung tumors. Drugs or small molecules that stabilize or activate mitochondrial pathways involving HCCS could potentially enhance responsiveness to current therapies.13,54,64
Limitations and future perspectives
This study has several limitations. First, the study was based on in silico analyses of publicly available datasets, which are subject to dataset heterogeneity and may not fully capture relevant clinical or biological confounders. Second, the findings are correlative and in silico in nature, and the absence of functional or experimental validation precludes causal inference. Future studies using CRISPR-based manipulation of HCCS in lung cancer models are needed to clarify its biological role. In addition, associations between HCCS methylation and expression require confirmation through integrative multi-omics analyses. We also note that multiple-testing correction was not applied due to the exploratory design, and survival stratification relied on a median expression cutoff, which may not represent the optimal prognostic threshold. These limitations should be considered when interpreting the results.
Conclusion
In summary, this integrative bioinformatics analysis identifies HCCS as a robustly dysregulated and prognostically significant gene in lung cancer, validated across TCGA, meta-analysis of 18 cohorts, and multiple GEO datasets. Despite its elevated expression in tumors, low HCCS expression predicts worse survival, suggesting a protective or tumor-suppressive role linked to mitochondrial homeostasis. Promoter hypermethylation in LUAD and associations with immune infiltration further emphasize the multifaceted biological role of HCCS in tumor progression. Collectively, these findings establish HCCS as a promising prognostic biomarker and potential therapeutic target in lung cancer. Functional and translational studies are now warranted to validate its mechanistic involvement and clinical applicability.
Supplemental Material
sj-docx-1-cbm-10.1177_18758592261429264 - Supplemental material for Integrative bioinformatics analysis identifies HCCS as a prognostic and therapeutic biomarker in lung cancer
Supplemental material, sj-docx-1-cbm-10.1177_18758592261429264 for Integrative bioinformatics analysis identifies HCCS as a prognostic and therapeutic biomarker in lung cancer by Sm Faysal Bellah, Md Alim Hossen and S.M. Saker Billah in Cancer Biomarkers
Footnotes
Acknowledgments
We thank to our research team for their insightful discussions and contributions throughout the project.
Ethical statements
This study is a computational biology research study and is exempt from Institutional Review Board approval
Author contributions
Sm Faysal Bellah: Conceptualization, Methodology, Formal analysis, Result validation, Writing- original draft, Supervision. Md Alim Hossen: Methodology, Formal analysis, Result validation, Writing - review & editing. S M Saker Billah: Methodology, Formal analysis, Writing - review & editing.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.