
Abstract
ARID1A mutations are a common occurrence in colorectal cancer (CRC) cells, but clinical therapeutic options targeting this anomaly remain unavailable. The loss of ARID1A functionality compromises DNA damage repair processes, potentially causing cancer cells to rely more heavily on PARP-dependent DNA repair pathways to preserve genomic integrity, thereby making them susceptible to PARP inhibitor (PARPi) therapy. To evaluate the suitability of PARPi treatment for CRC patients with ARID1A sufficiency (ARID1A+) and ARID1A deficiency (ARID1A-), our study enrolled 80 patients who had undergone surgical treatment for primary CRC. Surgical specimens underwent immunohistochemical examination to assess ARID1A protein expression. The study explored correlations between ARID1A expression loss and clinicopathological characteristics. Moreover, primary CRC cells were isolated through enzymatic digestion and validated using the colorectal carcinoma marker CK20. Subsequently, PARPi sensitivity was investigated in untreated ARID1A+ and ARID1A- CRC patients using an ATP-tumor chemosensitivity assay (ATP-TCA). Additionally, we confirmed the efficacy of PARPi in these primary CRC cells through clone formation and assessed its impact on cell cycle dynamics, apoptosis, and DNA damage repair signaling pathways using immunofluorescence and flow cytometry. The results demonstrated that the ARID1A- group displayed greater sensitivity to PARPi compared to the ARID1A+ group. PARPi treatment led to increased tumor cell death in the ARID1A- group. Mechanistically, ARID1A deficiency resulted in cell cycle abnormalities, particularly G2/M phase arrest, which was further exacerbated by PARPi treatment. Furthermore, PARPi treatment significantly increased the number of RAD51 foci in ARID1A- cell lines. In conclusion, our study highlights the potential of PARPi as an effective therapeutic option for ARID1A- CRC patients.
Introduction
Colorectal cancer (CRC) remains a predominant cause of cancer-related mortality worldwide, highlighting the pressing need for the development of innovative therapeutic strategies.1,2 The advent of precision medicine has ushered in a new era for cancer treatment, with poly(ADP-ribose) polymerase (PARP) inhibitors emerging as a promising class of targeted therapy.3,4 PARP inhibitors exploit synthetic lethality, a concept where the inhibition of PARP enzymatic activity leads to cell death in the context of specific genetic deficiencies.5,6 This therapeutic approach has gained substantial traction, particularly in cancers harboring deficiencies in DNA damage repair (DDR) mechanisms, such as BRCA1/2 mutations. However, the applicability of PARP inhibitors beyond these mutations remains an area of active investigation. 7
The chromatin remodeling complex plays a crucial role in maintaining genomic stability by modulating DNA repair processes. ARID1A, a key component of the SWI/SNF chromatin remodeling complex, is frequently mutated or lost in various cancer types. 8 ARID1A mutations are found in approximately 10%–15% of CRC patients.9,10 The loss of ARID1A impairs homologous recombination (HR), a high-fidelity DDR pathway, and has been proposed to create a vulnerability that can be targeted therapeutically. 11 Notably, recent preclinical studies have highlighted the potential of exploiting ARID1A deficiency to sensitize cancer cells to PARP inhibitors, a phenomenon that may extend the clinical utility of these drugs beyond BRCA-mutated cancers. 11
In CRC, the loss of ARID1A expression has been found in a lot of cases. This loss has been associated with various clinicopathological features and a poor prognosis, suggesting its critical role in CRC pathogenesis and progression. Given that ARID1A loss impairs HR, CRC cells harboring this deficiency may exhibit increased sensitivity to PARP inhibitors due to their increased reliance on PARP-mediated DNA repair pathways. 8 This synthetic lethality can lead to the accumulation of DNA damage, ultimately resulting in cell death. This approach aligns with the principle of precision oncology, where the selection of targeted therapies is based on the specific genetic alterations present within the tumor. 12
The aim of this study was to investigate the role of PARP inhibitors in ARID1A mutant colon cancer patient samples and to evaluate the effects of PARP inhibitors on DNA repair gene expression and cell cycle changes to ultimately improve the clinical use of PARP-based inhibitors.
Method
Patient sample collection
Fresh CRC tissue were obtained following surgical colorectal resection from 80 consented patients with no prior chemotherapy or radiotherapy exposure. Tumor stages were assessed using the TNM system as set by the American Joint Committee on Cancer (AJCC). Pathological categorization of each growth adhered to the classification standards of the World Health Organization.
Ethical considerations
Written informed consent was obtained from all participants, agreeing to the anonymous sharing of data. The independent ethics committee of MianYang Fulin Hospital in China granted approval for the study (IRB ID: TJ-C20220526), and which was conducted in line with the ethical principles of the World Medical Association Declaration of Helsinki. Each tissue sample was harvested within one-hour post-surgery. Part of each sample was allocated for the ATP-TCA test in the lab, while the remaining tissue was preserved in formalin for IHC processing.
Clonogenic survival assay
Cells were seeded in triplicate in 6-well culture dishes with an area of 9.6 cm2. They were allowed to adhere for 4–6 h before being exposed to PARPi, after which they were cultured for another 10–14 days at 37 °C with 5% CO2. The cells were then fixed and dyed with a mixture of 96% ethanol and 15% Giemsa, and finally rinsed with distilled water.
Immunohistochemistry (IHC) analysis and evaluation
The presence of ARID1A protein in 80 primary CRC specimens was investigated through IHC as detailed in prior reports.13,14 In summary, each tissue slice was subjected to deparaffinization, rehydration, and endogenous peroxide blocking, with antigen retrieval, followed by an incubation period with an ARID1A (PSG3) antibody (sc-32761, Santa Cruz Biotechnology, Inc., CA, USA) at 4 °C overnight. The sections were then washed in PBS mixed with 0.1% Tween-20, incubated for 30 min with a secondary antibody targeting mouse antigens, and then treated with streptavidin-HRP. Color was developed using Diaminobenzidine tetrahydrochloride, with hematoxylin used for subsequent counterstaining. Two independent evaluators, unaware of the patients’ clinical and pathological details, reviewed and rated all the stained tissue sections. Over 1000 cancer cells per slide were analyzed to assess staining intensity and the proportion of stained cells. A positive ARID1A expression was determined if over 60% of cells were stained, while a negative expression was indicated by 60% or fewer stained cells.
Immunofluorescence and Western blot
Cells cultured on coverslips were fixed using 4% paraformaldehyde for 20 min, followed by permeabilization with 0.2% Triton X-100 in PBS for 10 min. After blocking, the coverslips were incubated overnight at 4 °C with primary antibodies, including anti-53BP1(#4937S, 1:1000, Cell Signaling Technology), anti-RAD51 (PC130, 1:500, Merck Millipore). Following multiple washes with 0.1% Triton X-100 in PBS, coverslips were treated with Alexa Fluor 488-labeled anti-mouse or Alexa Fluor 594-labeled anti-rabbit secondary antibodies (#A11001, 1:500, Invitrogen) for one hour. Nuclei were stained with DAPI. Coverslips were mounted using fluorescence mounting medium (Agilent Dako, Santa Clara, CA, USA). Image analysis was performed using fluorescent microscope imager Z1 (Zeiss). For Western blot, Proteins separated by SDS-PAGE were transferred onto PVDF membranes. For detection, the rabbit anti-CK20 antibody was applied at a 1:2000 dilution, while anti-GAPDH antibody served as a loading control. The membranes were incubated for 2 h at room temperature before being washed with TBST (0.01% Tween 20 in tris-buffered saline). Subsequently, HRP-conjugated anti-rabbit antibodies (Pierce, Rockford, IL) diluted 1:1000 were used for a 1-h incubation at room temperature. Chemiluminescence (Arlington Heights, IL) was employed to visualize CK20 and GAPDH bands.
ATP-tumor chemoresistance assay (ATP-TCA)
The ATP-TCA was carried out in line with methods described in a previous publication. 8 Solid tumor tissues were finely chopped and digested with collagenase throughout the night (1.5 mg/ml, Sigma, Poole, UK; C8051). Following this, a Ficoll-Hypaque solution (Sigma; 1077-1) was used to remove excessive debris and red blood cells, after which cells were suspended in a complete medium devoid of serum (CAM; DCS Innovative Diagnostik Systeme, Hamburg, Germany) supplemented with gentamicin, penicillin–streptomycin, metronidazole (Rhône Poulenc Rorer, Eastbourne, UK), and amphotericin B (Sigma). Cell viability and counts were determined via trypan blue exclusion. The cell suspensions were then prepared at a density of ∼200,000 cells/ml for solid tumors and ∼100,000 cells/ml for malignant effusions. Polypropylene, round-bottomed 96-well plates (Corning-Costar, High Wycombe, UK) were arranged with CAM and inhibitors at six dilution levels (6.25–200%) in triplicate. The range for the therapeutic drug concentration (TDC) was established based on prior pharmacokinetic and biological response studies. All drug solutions were prepared and stored following the manufacturer's protocols. Drug dilutions were made from freshly prepared solutions, going up to 800% of the TDC. Drug combinations were tested by adding each drug at 800% of their TDC. Reference rows on each plate included a medium-only control (MO) with CAM without the drug, and a maximum inhibitor concentration (MI) to achieve full cell kill and a zero ATP reading. After a 6-day incubation at 37 °C, with 5% CO2 and 100% humidity, cells were lysed using the detergent-based Tumor Cell Extraction Reagent (DCS Innovative Diagnostik Systeme), and ATP levels were measured using a microplate luminometer with a luciferin–luciferase assay (MPLX; Berthold, Pforzheim, Germany).
Cell cycle
Cells were seeded into six-well plates at densities ensuring 70–80% confluence at the time of examination. After settling, the cells underwent treatment with 4.5 μM Olaparib for 48 h, followed by two PBS washes, trypsinization, and centrifugation at 800×g for 5 min. The cells were then fixed in 70% ethanol for 2 h at 4 °C. After ethanol removal, the cell pellet was washed twice with ice-cold PBS. The pellet was then suspended in PBS containing 50 μg/ml RNase at 37 °C for 30 min and subsequently stained with 50 μg/ml propidium iodide in darkness at 4 °C for 30 min. Flow cytometric analysis was conducted using a BD FACSCalibur flow cytometer (BD Biosciences, San Diego, CA, USA), and the data were processed using ModFit software (Verity Software House, Inc., Topsham, ME, USA)
Statistical analysis
Data are presented as mean ± standard deviation. Statistical analyses were performed using SPSS 14.0 and SAS software (version 14.1, SAS Institute, Cary, NC, USA). The χ2 test was used to examine the relationship between ARID1A expression and clinicopathological features. Statistical significance between the studied groups was determined by analysis of variance (one-way ANOVA) followed by Bonferroni's selected comparisons test. Statistical significance was defined as a p-value less than 0.05.
Results
Relationship between ARID1A mutation and clinicopathological characteristics in colon cancer patients
Tissue samples obtained from 80 individuals diagnosed with CRC were subjected to an assessment of ARID1A expression (Figure 1A). As shown in Table 1, 9 of 80 tumors (11.25%) were ARID1A negative. The clinical and pathological characteristics of the patients have been succinctly outlined in Table 1. It is noteworthy that the loss of ARID1A expression exhibited no correlation with gender, age, tumor location, TNM stage, or tumor size. Nonetheless, a statistically significant disparity in ARID1A expression was evident in relation to pathological differentiation (P < 0.05). The results indicate that ARID1A-negative CRC patients have poorly differentiated tumors.
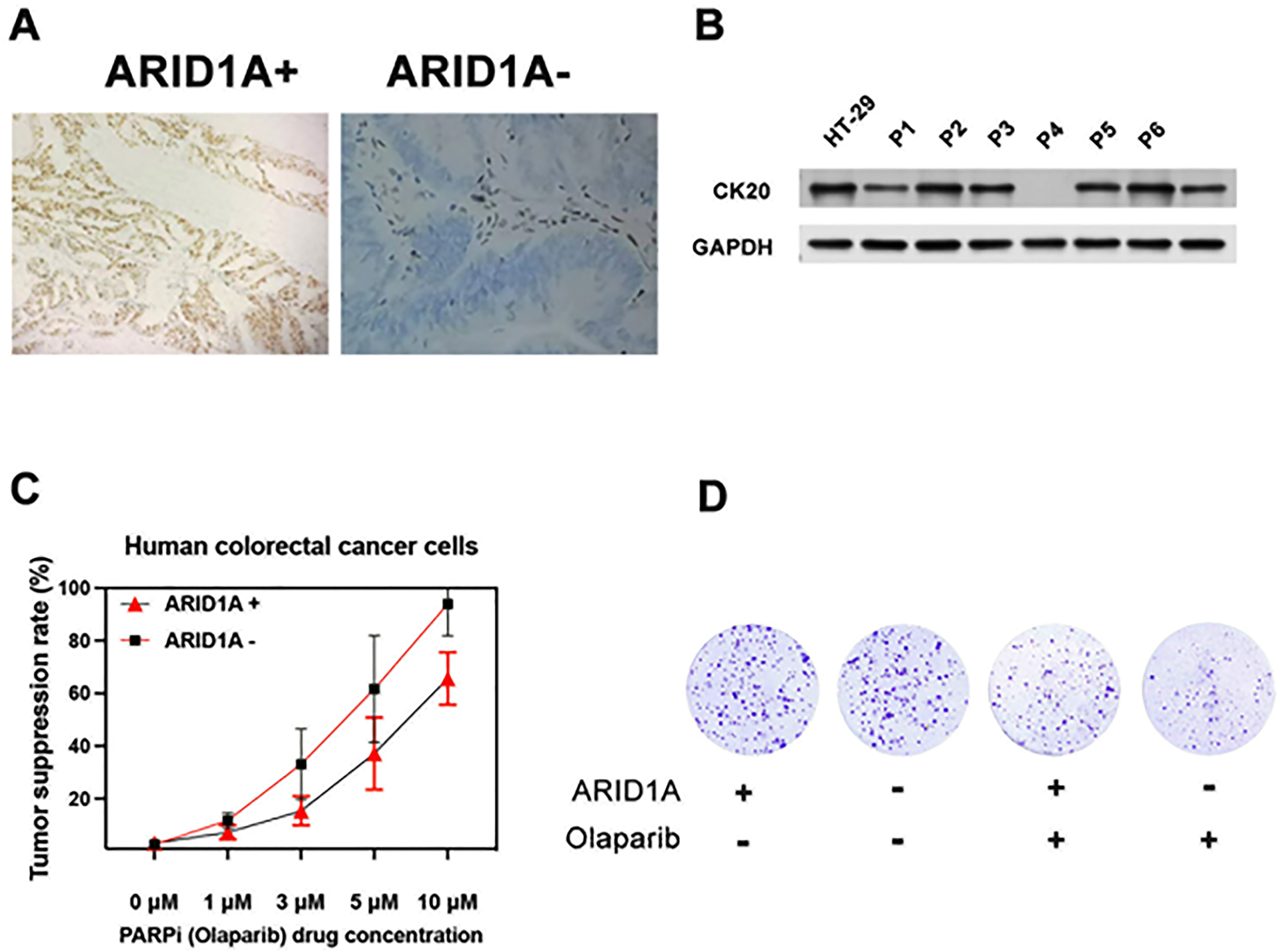
Effect of PARPi on ex vivo explants from CRC patients. (A) Illustration of immunohistochemistry (IHC) results showing the presence or absence of ARID1A expression in CRC clinical tumor. (B) Analysis of CK20 expression in primary CRC cells via Western blotting; HT-29 served as positive control and GAPDH served as a loading control. (C) Evaluation of the impact of the PARP inhibitor Olaparib on ex vivo CRC patient cells using the ATP-Tumor Chemosensitivity Assay. The assay measured ATP activity in ex vivo cells treated with Olaparib at concentrations ranging from 0 to 10 nM in CRC patient cells with (+) and without (−) ARID1A expression. (D) Assessment of the growth-inhibitory effects of Olaparib on both ARID1A+ and ARID1A- Ex Vivo CRC cells through a clonogenic assay. All experiments were repeated three times.
Relationship between ARID1A mutation and clinicopathological characteristics in colon cancer patients.
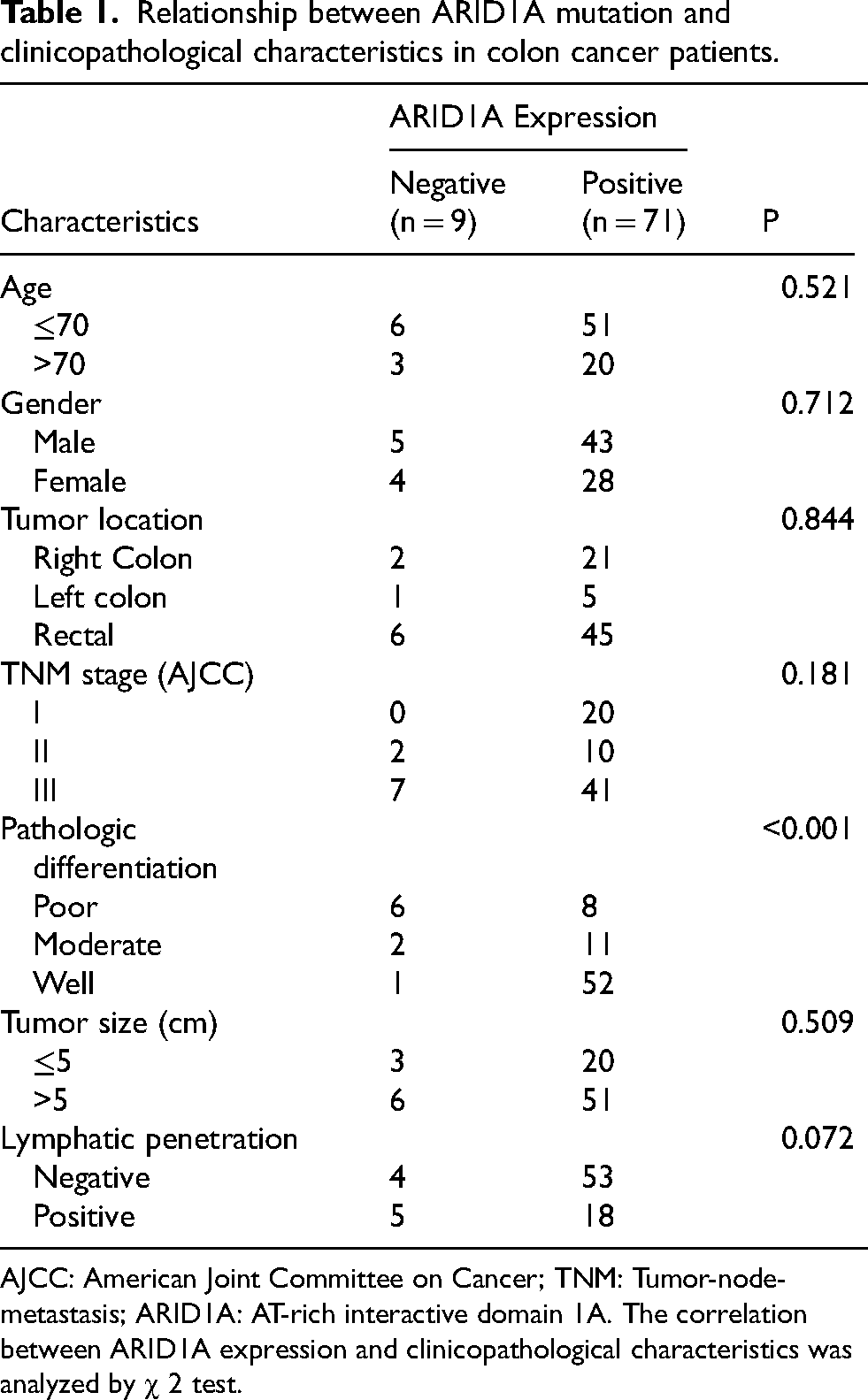
AJCC: American Joint Committee on Cancer; TNM: Tumor-node- metastasis; ARID1A: AT-rich interactive domain 1A. The correlation between ARID1A expression and clinicopathological characteristics was analyzed by χ 2 test.
Measurement of PARP inhibitor's impact on ex vivo tumor cell growth inhibition
The impact of PARP Inhibitor on CRC Ex Vivo Tumor Cell was assessed in an ATP-based tumor chemosensitivity assay (ATP-TCA). 68 of 80 successfully underwent the ATP-TCA analysis, while 12 samples were excluded due to insufficient cell quantities or weak expression of the colon cancer marker CK20 15 (Figure 1B, Supplemental Figures S1 and S2). These 68 cases, comprising 7 ARID1A-negative and 61 ARID1A-positive colorectal cancer samples, were utilized to establish a Olaparib dose-response curve employing the ATP-TCA technique (as depicted in Figuer 1C, with enhanced cell viability on the y-axis). The findings distinctly indicated that the ARID1A-negative group exhibited markedly higher sensitivity to Olaparib than the ARID1A-positive group, as evidenced by IC50 values of 4.53 ± 2.6 nM and 8.5 ± 3.3 nM for ARID1A-negative and positive groups, respectively. We next confirmed this anti-proliferative effect of Olaparib in ARID1+ and ARID1A - Ex Vivo CRC cells by clonogenic assay under incubation of 14 days with continuous exposure to Olaparib (Figure 1D) (Table 2).
The surviving fraction of PARP inhibitor on ARID1A+ and ARID1A- Ex vivo CRC cells colon cancer cell lines.
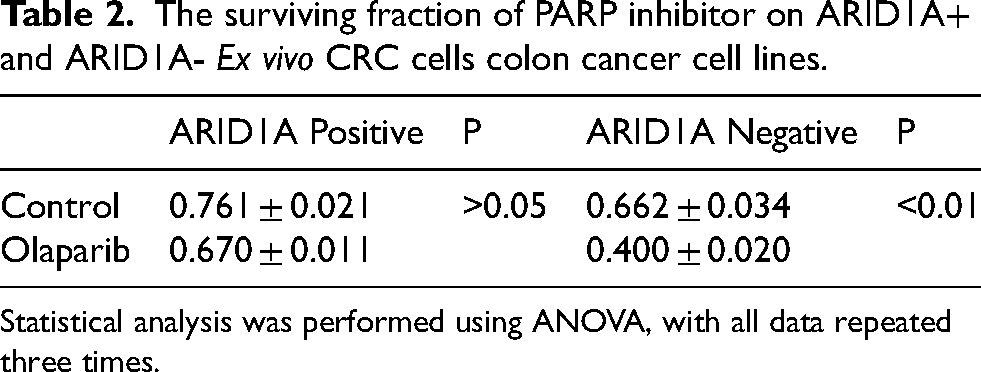
Statistical analysis was performed using ANOVA, with all data repeated three times.
Evaluating the effects of PARP inhibitor on double-strand breaks (DSBs) and cell cycle arrest in ARID1A-mutated ex vivo tumor cells
We explored the cell cycle-specific impact of PARPi on ARID1A- CRC by conducting a cell cycle assay on our Ex Vivo colon cancer cells. The results depicted in Figure 2A indicate that ARID1A- plus PARPi led to a higher percentage of cells in the G2 phase, as opposed to other groups (P < 0.05, respectively). To unravel the underlying mechanism of PARP Inhibitor in ARID1A- CRC cell lines, we detected DSBs signal 4 h after PARP inhibitor treatment. We firstly assessed RAD51 foci formation as an indicator of homologous recombination repair (HRR). PARPi significantly increased the RAD51 foci in the ARID1A- group, in comparison to other groups (P < 0.05, respectively). Subsequently, we checked 53BP1 foci formation as a marker for non-homologous end-joining (NHEJ) activity post DNA damage. The combination of PARPi and ARID1A- also impacted the formation of 53BP1 foci in cell lines (Figure 2B).
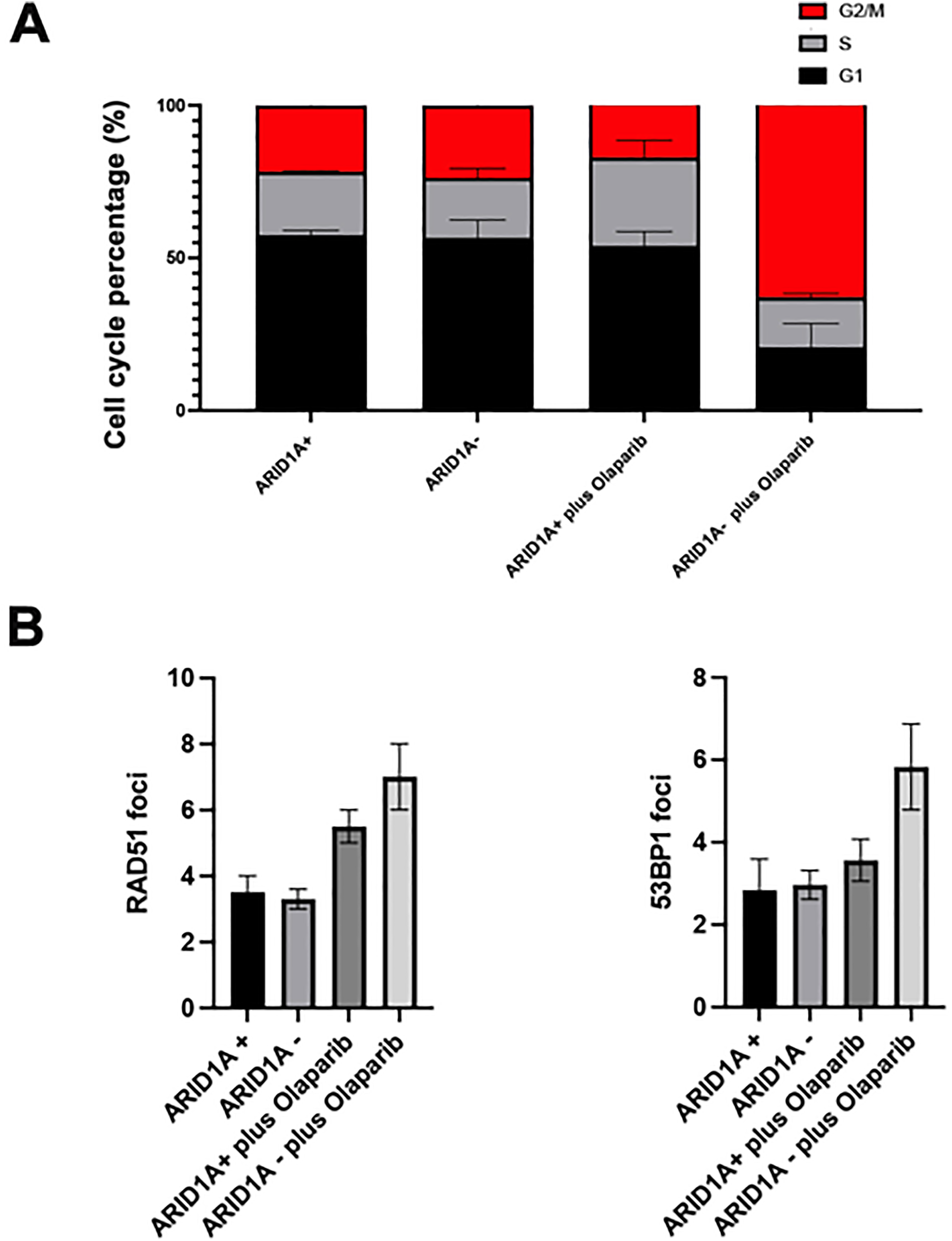
Effects of PARP inhibitors on double-strand breaks (DSBs) and cell cycle arrest in ARID1A-mutated ex vivo tumor cells. (A) Effect of PARP Inhibitor on cell cycle progression; (B) Statistical evaluation PARP Inhibitor on RAD51 and 53BP1 foci formation in cells 4 h after PARP inhibitor treatment. Mean values are shown with standard deviation from 3 independent experiments.
Disscussion
The frequent mutation of ARID1A in colorectal cancer (CRC) cells presents a critical area of investigation, especially in the context of developing targeted therapeutic approaches.16–18 Current clinical options do not directly address the deficits caused by ARID1A mutations, underscoring the necessity for innovative treatments. The loss of ARID1A is known to impair DNA damage repair mechanisms in cancer cells, which in turn increases their dependency on PARP-dependent DNA repair pathways. 11 This reliance potentially renders ARID1A-deficient cells more susceptible to PARP inhibitor (PARPi) therapy.
In our study, the assessment of ARID1A expression in 80 primary CRC patients showed that 9 tumors (11.25%) were ARID1A negative. The loss of ARID1A expression did not correlate with various clinicopathological characteristics such as gender, age, tumor location, TNM stage, or tumor size. However, a significant association was noted with pathological differentiation. This suggests that ARID1A mutation it could be crucial in understanding tumor biology, especially in relation to differentiation. Our results are consistent with other findings that ARID1A protein loss is associated with poor pathological classification in colorectal cancer patients. 19
We further assessed the impact of PARP inhibitor on Ex Vivo CRC cell growth. Using an ATP-based tumor chemosensitivity assay (ATP-TCA), it was found that cells from ARID1A-negative tumors were more sensitive to the PARP inhibitor compared to ARID1A-positive ones. This was reflected in the distinct IC50 values for both groups. Additionally, the anti-proliferative effect of PARP inhibitor, Olaparib, was confirmed in both ARID1A-positive and negative ex vivo CRC cells through a clonogenic assay. The sensitivity of ARID1A-negative cells to PARP inhibitors might be indicative of an inherent vulnerability in these cells, which can be exploited targeted therapeutically. Previous research has indeed identified the targeted action of PARP inhibitors in ARID1A-mutated models, but the majority of these studies were conducted using either in vitro cell lines or animal models.11,20 The direct application of clinical patient cells in this study not only corroborates these earlier findings but also extends them by providing a more clinically relevant context. This is crucial for the development of personalized medicine strategies, as it underscores the potential of using patient-specific tumor profiles to guide treatment decisions.
Our study also investigated the effects of PARP inhibitors on DNA repair mechanisms in ARID1A-mutated cells. It was observed that treatment with PARP inhibitors led to an increased number of cells in the G2 phase of the cell cycle, suggesting PARP inhibitors might induce cell cycle arrest. Cell cycle arrest in the G2 phase can make cancer cells more susceptible to damage by radiation therapy.21–24 Moreover, an increase in RAD51 foci formation in the ARID1A-negative group was noted upon treatment with PARPi. RAD51 is a key protein in homologous recombination repair (HRR), and its increased formation indicates enhanced HRR activity. Furthermore, the formation of 53BP1 foci, a marker for non-homologous end-joining (NHEJ) repair post-DNA damage, was also influenced by the combination of PARPi and ARID1A mutation. Based on our results, we hypothesize that ARID1A+ cells may possess a relatively intact homologous recombination (HR) repair pathway, allowing them to resolve DNA damage caused by Olaparib without triggering a G2 arrest. In contrast, ARID1A-deficient cells often have impaired HR repair, making them more sensitive to PARP inhibitors and more likely to undergo cell cycle arrest. Our findings align with earlier results and propose that combining PARP inhibitors with radiotherapy could increase the sensitivity of tumor cells to radiation, especially considering the role of radiotherapy as a vital measure in cancer treatment.25,26 The underlying mechanism of this synergy lies in the ability of PARP inhibitors to generate replication-associated double-strand breaks (DSBs), which require homologous recombination (HR) for accurate repair. Given that ARID1A loss impairs HR, colorectal cancer (CRC) cells with this deficiency may exhibit increased sensitivity to PARP inhibitors due to their reliance on alternative repair mechanisms, such as error-prone alternative end-joining (alt-EJ).27,28 This dual effect—compromising DNA repair fidelity and inducing cell cycle arrest—may enhance the effectiveness of radiotherapy, leading to improved tumor control. Although the results are promising, there are a few limitations to our study. First, our study groups were relatively small, with a limited number of patients. Therefore, large-sample multicenter studies will be required to validate our data set. Additionally, despite the importance of ex vivo experiments in clinical drug evaluation, in vivo experiments are still necessary for further validation.
In summary, our study underscores the potential of PARPi as an effective treatment modality in ARID1A-deficient CRC. These findings could pave the way for more personalized cancer therapies, targeting specific genetic alterations in CRC. Future research should focus on further elucidating the mechanisms underlying the sensitivity of ARID1A- CRC cells to PARPi and exploring the potential of combining PARPi with other therapeutic agents to enhance treatment efficacy.
Supplemental Material
sj-tif-1-cbm-10.1177_18758592251317873 - Supplemental material for Evaluating the efficacy of PARP inhibitor in ARID1A-deficient colorectal cancer: A ex vivo study
Supplemental material, sj-tif-1-cbm-10.1177_18758592251317873 for Evaluating the efficacy of PARP inhibitor in ARID1A-deficient colorectal cancer: A ex vivo study by Rui Zong, Ping Zhou, Shaojie Qin, Jie Li, Shan Xu, MingWei Kang and Yuping Zhang in Cancer Biomarkers
Supplemental Material
sj-tif-2-cbm-10.1177_18758592251317873 - Supplemental material for Evaluating the efficacy of PARP inhibitor in ARID1A-deficient colorectal cancer: A ex vivo study
Supplemental material, sj-tif-2-cbm-10.1177_18758592251317873 for Evaluating the efficacy of PARP inhibitor in ARID1A-deficient colorectal cancer: A ex vivo study by Rui Zong, Ping Zhou, Shaojie Qin, Jie Li, Shan Xu, MingWei Kang and Yuping Zhang in Cancer Biomarkers
Footnotes
Acknowledgements
The authors thank the reviewers for their helpful comments on this article. The authors are also grateful to Mogo Editing for polishing and revising the language.
Ethics approval and consent to participate
The present study was approved by the ethics committee of MianYang Fulin Hospital in China (IRB ID: TJ-C20220526).
Patient consent for publication
Not applicable.
Author contribution statement
Conception: R.Z, YP.Z, KM.W and S.X; Interpretation or analysis of data: J.L, P.Z, S.X, KM.W and R.Z; Preparation of the manuscript: P.Z and SJ.Q; Revision for important intellectual content: YP.Z, R.Z and S.X; Supervision: YP.Z and S.X.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.