
Abstract
The influence of different parameters (solid–liquid ratio, initial pH, initial Cu concentration and anion type) on the cementation of aqueous copper (Cu) with nanoscale zerovalent iron (nZVI) has been studied. The work has been established to study both the influence such parameters have on the kinetics and efficacy of the cementation process but also the physicochemical composition of resultant Cu-bearing products. The nZVI exhibited high Cu removal capacity (maximum removal 905.2 mg/g) due to its high surface area. X-ray diffraction determined the most common Cu-bearing precipitates were Cu2O, CuCl2 and Cu2(OH)3Cl for solutions containing Cl− counterions (CuCl2 salt precursor), while Cu0 and Cu2O were the most common phases for those containing
Introduction
Copper (Cu) remains a valuable commodity due to its widespread use in a range of different industrial and domestic products and processes (electric circuitry, transportation, metallurgy, construction, medicine, petroleum refining, etc.). The concurrent global transition towards a circular economy means that there is strong current demand for the development of new processes that can be used to recover Cu during hydrometallurgical processing. Even more pressing is the need to find economically viable means in which to capture aqueous Cu when it is present as a component of wastewaters and effluents, for example, those derived from mining, processing and disposal of Cu. 1 Furthermore, whilst Cu is an essential trace element exposure to excess Cu is known to cause a wide range of health and environmental issues, with permissible limits given by the World Health Organisation (WHO) for dissolved Cu in drinking water currently 2 mg/L 2 and contamination of water resources with Cu a significant global environmental problem. Therefore, the development of reliable and cost-effective methods for the removal/capture of Cu from wastewaters and effluents is an important technical challenge for environmental protection and prevention of Cu ‘leakage’ from the materials loop.
Conventional methods for the removal of Cu ions from solution include adsorption, precipitation and ion exchange. Among them, chemical precipitation (often by lime or sulphide salts) has become one of the most widely applied due to its low cost. However, the relatively high solubility of Cu2+ dictates that relatively high quantities of reagents are often required, resulting in large quantities of sludge, 3 with the disposal of such sludge adding an additional cost and environmental concern. 3
Rather than creating secondary waste (which requires disposal), the direct formation of functional Cu-based materials and products would be much more beneficial, because it would enable dissolved Cu to be efficiently recycled with minimal unwanted by-products. Cementation is a hydrometallurgical process that involves the precipitation of a dissolved metal ion by another more electropositive metal through an electrochemical reaction. Various researchers have studied cementation reactions for different metal ions such as gold (Au), aluminium (Al) and zinc (Zn). Cu is considered to be one of the most valuable metals which can be recovered by this technique, with a number of studies documenting the efficient cementation of Cu onto Fe0. 4 Indeed, before being superseded by solvent extraction methods, cementation was the principal method that Cu was recovered during hydrometallurgical processing of Cu ores. While the use of Fe0 for Cu cementation in mining is therefore now redundant, the process remains highly suitable for Cu removal in effluent treatment applications due to the relatively low cost of zerovalent iron (ZVI) as a reagent.
In recent years, nanoscale zerovalent iron (nZVI) has gained much attention for its use in a wide range of water treatment applications due to its unique properties, including extremely high surface area to volume ratio and an ability to be injected into the subsurface as a colloidal suspension, 5 –8 The high efficacy of nZVI for the removal of Cu2+ has been reported in a number of empirical studies, 9,10 ; however, very little information currently exists on the physicochemical properties of the resultant Cu particles (particle size, particle shape, crystallinity, etc.) when removed from solution by nZVI. Indeed, if the cementation reaction of aqueous Cu with nZVI could be harnessed to produce discrete and pure Cu nanoparticles, then the value of the Cu in the aqueous waste stream could significantly increase when compared to extracting Cu simply as a bulk scale material (e.g. a plate or nugget). Cu-based nanomaterials have received much attention due to their unique catalytic, optical, electrical and antifungal/antibacterial applications. For example, Cu2O is a p-type semiconductor with a direct bandgap of 2 eV, which makes it a highly promising material for the conversion of solar energy into electrical or chemical energy. 11 Moreover, monometallic and Cu oxide nanoparticles have received much attention due to their unique antimicrobial properties while also being significantly lower cost than more conventional Ag-based (nano)materials. 12 Recent research has also demonstrated that nanostructured copper cathodes (namely nanoscale Cu2O) are among the most efficient and selective catalysts to date for the electrochemical reduction of carbon dioxide into multicarbon products. 13 Nanoscale Cu chloride and Cu chloride hydroxide forms have also received much attention in recent years due to their unique physicochemical properties. Among these materials, paratacamite (Cu2(OH)3Cl) is one of the most thermodynamically stable alkaline copper chloride phases and is known to exhibit unique applications in a wide range of sectors including catalysis, 14 corrosion prevention, 15 agriculture 16 and the uptake and storage of hydrogen. 17 Moreover various forms of cupric and cuprous chloride are used as catalysts in a wide range of reactions including the Wacker process, the production of chlorine by oxychlorination and the synthesis of numerous different chlorinated organic compounds.
In this work, the cementation reaction between nZVI and aqueous Cu has been investigated under a range of different chemical conditions. Various researchers have studied the impact of different experimental parameters on the cementation reaction between aqueous Cu and bulk scale Fe0 (i.e. micron scale particles or larger); however, similar experiments using nZVI are currently lacking in the literature. This work has therefore been established to provide this fundamental mechanistic data but also provide specific insight into the physiochemical composition of Cu-bearing (nano)material products, including data on the extent at which the composition of such (nano)materials (metallic, oxide, hydroxide, chloride, etc.) is influenced by the chemistry of the Cu-bearing precursor solution.
Materials and methods
Synthesis of nZVI particles
Pure nZVI was synthesised following the methodology first described by Glavee et al., 18 and then adapted by Wang and Zhang 19 ; 7.65 g of FeSO4·7H2O was dissolved in 50 mL of Milli-Q water (>18.2 MΩ cm), and then a 4 M NaOH solution was used to adjust the pH to 6.8. The salts were reduced to nZVI by the addition of 3.0 g of sodium borohydride (NaBH4). The nanoparticle product was isolated from the aqueous phase via centrifugation (Sigma 3-16L centrifuge, 4000 r/min (3077 × g) for 240 s), rinsed with absolute ethanol (approximate ratio of 50 mL/g of nZVI) and then centrifuged (Sigma 3-16L centrifuge, 4000 r/min (3077 g) for 240 s). This step was then repeated three more times. The nanoparticles were dried in a vacuum desiccator (approximately 10−2 mbar) for 48 h and then stored in an argon filled (BOC, 99.998%) MBraun (MB-acryl GB 2202-P-VAC) glovebox until required.
Experimental procedure
Four 200 mL solutions were synthesised using reagent grade CuCl2 salt (Sigma Aldrich, 222011) comprising Cu at 50, 100, 500 and 1000 mg/L. A further three 200 mL solutions were also synthesised using CuCl2 salt with Cu at 1000 mg/L. The pH of these solutions was adjusted to pH 2.0, 3.0 and 4.0 using a few drops of 0.1 and, where appropriate, 1 M concentration HCl. The same process was then conducted but using CuSO4 salt (Sigma Aldrich, C1297) and 1 M H2SO4 for pH modification. All solutions were stored in 250-mL clear soda lime glass jars (Fisher Scientific, 11704329) and left for 24 h to equilibrate before commencing each experiment. The nZVI was added to each batch system at a concentration of 0.1 g/L and immediately sonicated for 240 s using an ultrasonic bath (Grant, XB3). Each batch system was then placed on the benchtop and periodically tested for pH and Eh. The pH probe was calibrated prior to each measurement. The measured oxidation/reduction potential (ORP) values were converted to Eh values by subtracting the difference between the measured ORP of the reference solution and the theoretical ORP of the reference solution (220 mV). Nanoparticle suspensions were extracted from each batch system at the same time. Prior to sampling each jar was gently agitated to ensure homogeneity and then 10 mL was removed using an auto-pipette. The extracted suspensions were then centrifuged at 4000 r/min (3077 × g) for 240 s. The supernatant was then filtered through a 0.2 µm cellulose acetate filter and acidified (HNO3, 2% by volume) for inductively coupled optical emission spectrometry (ICP-OES) analysis. The nanomaterial plug (at the base of the centrifuge) was then washed using absolute ethanol (approximate ratio of 5 mL/mg of nZVI) and then centrifuged at 4000 r/min (3077 × g) for 240 s again. The supernatant was then decanted. The resultant solid material was then pipetted onto a glass optical microscope slide (Agar Scientific, G251P) and an Au-coated holey carbon film (TAAB, C062/G) for X-ray diffraction (XRD) and high-resolution transmission electron microscopy (HR-TEM) analysis, respectively. Samples were dried in a vacuum chamber at <1 × 10−2 mbar for a minimum of 2 h prior to analysis. All sorption–desorption experiments were conducted at room temperature (measured to be 20 ± 1°C) and ran as duplicates, with the average data used to create the figures/tables displayed herein.
Analysis techniques
X-ray photoelectron spectroscopy (XPS) spectra were collected using a Thermo Scientific K-Alpha+ XPS spectrometer. Spectra were collected at a pass energy of 40 eV for narrow scans and 150 eV for survey scans with a 0.1 and 1 eV step respectively. Charge neutralisation was achieved using a combination of low-energy electrons and argon ions. Spectra were quantified in CasaXPS using Scofield sensitivity factors and an energy dependence of −0.6. Curve fitting was carried out using Pisces software (Dayta Systems Ltd, 2011) 20 with binding energy values of the recorded lines referenced to the adventitious hydrocarbon C1s peak at 284.8 eV. To determine the relative proportions of Fe2+ and Fe3+ in the sample analysis volume, curve fitting of the recorded Fe 2p3/2 photoelectron peaks was performed following the method of Scott et al. 21 The Fe 2p3/2 profile was fitted using photoelectron peaks at 706.7, 709.1, 710.6 and 713.4 eV corresponding to Fe0, Fe2+ octahedral; Fe3+ octahedral and Fe3+ tetrahedral. These parameters were selected on the basis that the surface oxide was assumed to be a mixture of wüstite and magnetite, as the oxide Fe2+ is in the same coordination with the surrounding oxygen atoms in both forms of oxide. Brunauer-Emmett-Teller (BET) surface area analysis was performed using a Quantachrome NOVA 1200 surface area analyser, with N2 as the adsorbent and following a seven-point BET method. Prior to analysis, the samples were degassed under vacuum (approximately 10−2 mbar) for a 24 h period at a temperature of 75°C. Samples were ran as triplicates with the average recorded. Inductively coupled plasma optical emission spectrometry (ICP-OES) analysis of aqueous samples (to determine Cu and Fe concentrations) was performed using a Perkin Elmer Optima 2100 DV ICP-OES. A Phillips Xpert Pro X-ray diffractometer with a CoKα radiation source was used for XRD analysis (generator voltage of 40 keV and tube current of 30 mA). XRD spectra were acquired between 2 h angles of 10–90°, with a step size of 0.02 and a 2 s dwell time. Data were fitted with using X-pert HighScore Plus software. HR-TEM analyses were performed using a JEOL JEM-2100 microscope at 200 kV. Energy dispersive X-ray (EDX) analysis and mapping was performed using Oxford Instruments X-MaxN analyzer and Aztec software. A beryllium sample holder was used to prevent any background Cu from being detected. Nanoparticle size distribution from TEM images was measured using ImageJ software (Java 1.6.0_24) with 50 nanoparticles analysed per sample. Samples were prepared for zeta potential analysis by synthesising 20 mL CuCl2 or CuSO4 bearing solutions with Cu at 1000 mg/L. To each batch system, nZVI was added at a concentration of 0.1 g/L. The nanoparticle suspensions were then sonicated for 240 s using an ultrasonic bath (Grant, XB3) and then left on the benchtop in the open laboratory. Each suspension was then centrifuged at 4000 r/min (3077 × g) for 240 s. The supernatant was then decanted, and 40 mL of absolute ethanol was added. The suspension was then centrifuged at 4000 r/min (3077 × g) for 240 s; 40 mL of a 0.1 M NaCl solution was then added. The zeta potential of the nanoparticles was then measured using a ZetaSizer Nano ZS instrument (Malvern Instruments Ltd). A refractive index of 1.86 and 0.46 was used for the nanoparticles formed from CuCl2 and CuSO4 solutions, respectively (corresponding to CuCl and Cu respectively) and an absorbance of 0.1. The pH was adjusted prior to each measurement using HCl or NaOH at 0.1 M concentrations.
Results and discussion
Characterisation of the unreacted nZVI
TEM analysis determined that the nZVI were spherical, generally within an approximate size range of 10–150 nm and an average diameter of 61 nm (Figure 1). Each individual nZVI particle was recorded to contain a discrete outermost layer (density contrast), which is attributed to be the presence of an oxide shell surrounding the Fe0 core. In addition, dark mottles were recorded within the metallic cores which indicate that individual particles are either polycrystalline or comprised of isolated metal crystals in an otherwise amorphous matrix. Individual nZVI particles were recorded as aggregated into chains and rings due to their high surface energy and magnetic properties. 22 BET surface area analysis determined that the surface area of the nZVI was 50.9 m2/g. A single diffraction peak at 52.381° 2θ was recorded using XRD and attributed to be the (110) lattice reflection of α-Fe0 (Figure 2). The peak is recorded as relatively broad which indicates that the nZVI are somewhat amorphous. XPS analysis recorded the outer surface of the nZVI to be comprised of a mixed valent (Fe2+/Fe3+) oxide (Figure 3) overlying a Fe0 core. Given the mean free path of Fe is equivalent to approximately five atomic layers, this detection of Fe0 in the XPS analysis volume therefore indicates that the oxide thickness is less than approximately 5 nm, which corresponds well with the aforementioned nZVI oxide thickness measurement using TEM. Results are summarised in Table 1 and concur with previous characterisation studies of nZVI, 23 –26

TEM images of the unreacted nZVI. TEM: transmission electron microscopy; nZVI: nanoscale zerovalent iron.
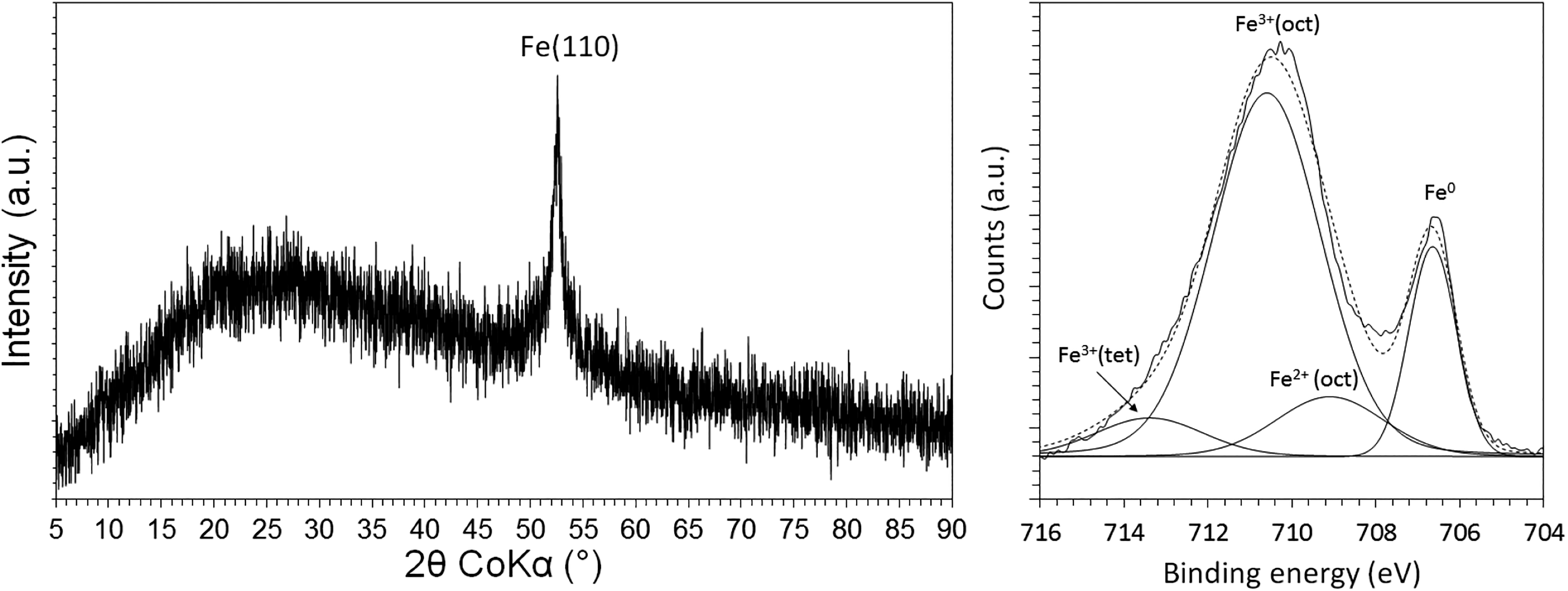
XRD (LHS) and curve fitted Fe 2p3/2 XPS spectra (RHS) for the unreacted nZVI. XRD: X-ray diffraction; nZVI: nanoscale zerovalent iron.

Eh and pH as a function of time for batch systems containing 50, 100, 500 and 1000 mg/L Cu for batch systems containing CuCl2 (top) and CuSO4 (bottom). An nZVI concentration of 0.1 g/L was used. nZVI: nanoscale zerovalent iron; Cu: copper.
Bulk and surface properties of the nZVI.a
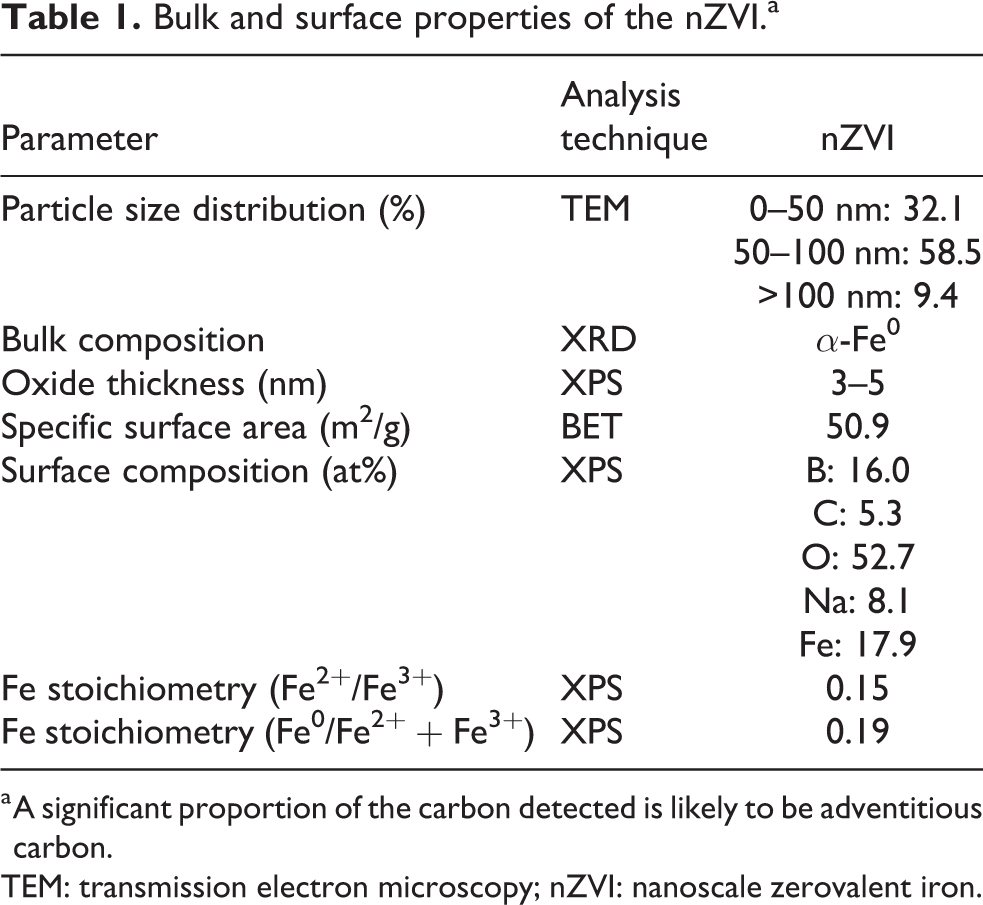
a A significant proportion of the carbon detected is likely to be adventitious carbon.
TEM: transmission electron microscopy; nZVI: nanoscale zerovalent iron.
Changes in pH and Eh
For all experimental systems, the addition of nZVI to the solutions resulted in a rapid decrease in Eh and a concurrent increase in pH (Figures 3 and 4). Eh minima were recorded within the first 2 h of reaction for all systems, while pH maxima were all recorded within the first 4 h of reaction. This behaviour is attributed to the rapid oxidation of the nZVI surfaces during their initial exposure to the Cu-bearing solutions, consuming dissolved oxygen (DO) and H+ and increasing the reduction potential of the system. Following this initial nZVI reaction period (e.g. <4 h), a recovery in pH and Eh was recorded in all systems, with reversal to near ambient (pre nZVI addition) conditions recorded for all systems recorded after 24 h.

Eh and pH as a function of time for batch systems containing 1000 mg/L Cu for batch systems containing CuCl2 (top row) or CuSO4 (bottom row) at a starting pH of 2.0, 3.0, and 4.0. An nZVI concentration of 0.1 g/L was used. nZVI: nanoscale zerovalent iron; Cu: copper.
No clear difference in changes in Eh and pH was recorded for batch systems with different starting Cu concentrations (Figure 3). In contrast, a marked difference was recorded for batch systems with different starting pH, with the greatest change typically recorded for the batch systems with the highest starting pH (Figure 4). For example, Eh minima were recorded as 245, 197 and 188 mV for the batch systems containing Cl− and with starting pH of 2, 3 and 4, respectively. Similarly, Eh minima were recorded as 362, 259 and 219 for the batch systems containing
Changes in the concentrations of Cu and Fe ions
In all systems, most significant Cu removal (and simultaneous Fe dissolution) was recorded during the initial stages of the reaction (typically <4 h; Figures 5
to 7). In general, the greatest removal of Cu from solution was recorded for systems containing the highest starting Cu concentrations, and greater Cu uptake was general recorded for solutions containing

Normalised Cu concentrations (C/C0) and absolute Fe concentrations (mg/L) as a function of time in each batch system (circle markers: solutions containing CuCl2, square markers: solutions containing CuSO4). From top to bottom: 50, 100, 500 and 1000 mg/L starting Cu concentration. An nZVI concentration of 0.1 g/L was used. Starting pH was 4.5–6 (see Figure 3). nZVI: nanoscale zerovalent iron; Cu: copper.

Normalised Cu concentrations (C/C0) and absolute Fe concentrations (mg/L) as a function of time in each batch system containing 1000 mg/L Cu (circle markers: the CuCl2 systems, square markers: the CuSO4 system). Top row: starting pH = 2.0; middle row: starting pH = 3.0; bottom row: starting pH = 4.0. An nZVI concentration of 0.1 g/L was used. nZVI: nanoscale zerovalent iron; Cu: copper.

Normalised Cu concentrations (C/C0) and absolute Fe concentrations (mg/L) as a function of time in each batch system containing 1000 mg/L Cu (circle markers: the CuCl2 systems, square markers: the CuSO4 system). An nZVI concentration of 1 g/L and an initial pH of 4.0 was used for each system. nZVI: nanoscale zerovalent iron; Cu: copper.
Starting pH exhibited a clear influence on Cu uptake, with greater total Cu removal generally recorded at higher pH. For example, maximum uptake of 784.1, 833.1 and 905.2 mg/g was recorded for CuSO4 solutions with a starting pH of 2.0, 3.0 and 4.0 respectively compared to 155.0, 298.9 and 321.1 for CuCl2 solutions, respectively. The standard reduction potential (E 0) of Cu/Cu2+ and Fe/Fe2+ couples is 0.34 and −0.44 V, respectively, and as such the removal of Cu onto nZVI is ascribed to the simultaneous oxidation (and dissolution) of Fe0 and chemical reduction of Cu2+ via the following cementation reaction:

Following this initial Cu uptake period, a slight increase (typically <10%) in aqueous Cu concentrations was recorded for all systems. This suggests that a proportion of the solid Cu that was removed from solution via cementation with nZVI was in a relatively reactive state and as such dissolved back into solution in the latter stages of the experiment concurrent with a reversal in both solution pH and Eh to ambient (pre nZVI addition) conditions. No clear correlation between the extent of Fe dissolution and the starting concentrations of CuCl2 or CuSO4 was recorded; however, a correlation was recorded with the starting pH of the solution, with greatest Fe dissolution typically recorded at low pH. After 24 h exposure of the Cu-bearing solutions to nZVI at a 0.1 g/L concentration, aqueous Fe concentrations were recorded to be 20.3, 30.3, 21.5 and 38.2 mg/L for batch systems containing Cl− as the counterion (50, 100, 500 and 1000 mg/L aqueous Cu starting concentrations respectively) and 26.3, 49.2, 79.0 and 74.5 mg/L for batch systems containing
XRD data
The XRD data demonstrate that Cu0 and Cu2O phases are the most common products for solutions containing CuSO4, while Cu2O, CuCl2 and Cu2Cl(OH)3 predominate for solutions containing CuCl2 (Figures 8 and 9). The only batch system to record a Fe-bearing XRD pattern was the system containing

XRD spectra recorded for nZVI extracted from solutions containing CuCl2 (top row) or CuSO4 (bottom row) at 50, 100, 500 and 1000 mg/L (from left to right). Spectra are for the nZVI extracted from each system after 0, 0.5, 1, 2, 4 and 24 h reaction time (stacked from bottom to top). An nZVI concentration of 0.1 g/L was used. Starting pH was 4.5–6 (see Figure 3). XRD: X-ray diffraction; nZVI: nanoscale zerovalent iron; Cu: copper.

XRD spectra recorded for nZVI extracted from solutions containing CuCl2 (top row) or CuSO4 (bottom row) at a starting pH of 3.0 (LHS) and 4.0 (RHS). Spectra are for the nZVI extracted from each system after 0, 0.5, 1, 2, 4 and 24 h reaction time (stacked from bottom to top). It was not possible to record XRD spectra for the systems with a starting pH of 2.0 due to acidic dissolution resulting in insufficient material being present. XRD: X-ray diffraction; nZVI: nanoscale zerovalent iron.
Overall the results demonstrate that in systems containing relatively low concentrations of Cl− (e.g. <100 mg/L), Cu2O is the dominant Cu-bearing product, with its formation likely via equation (2). In contrast, in systems containing

Using equation (2) Cu2+ is reduced to cuprite at an Fe to Cu molar ratio of 1:2, and this may explain (in part) the relatively high reduction capacity (e.g. >500 mg Cu per g nZVI) recorded herein.
TEM data
TEM images of the nZVI after 4 h reaction with the 1000 mg/L CuCl2 or CuSO4 solutions, along with EDX data of the four most common elements (by wt%) present, are displayed in Figures 10 and 11. Two distinct types of nanomaterial can be observed in both figures, hereafter referred to as ‘needles’ and ‘spherical’ nanoparticles, with the former comprised predominantly of Fe and O, and the latter comprised predominantly of Cu and O. Corroborating these data with the XRD data, it can therefore be stated that the needle-shaped nanoparticle are likely to be nZVI corrosion products (i.e. largely composed of amorphous iron (hydr)oxides), whereas the spherical particles are likely to be a mixture of Cu0 and Cu2O nanoparticles, which have formed via cementation ((1) and (2)). This result is significant because it proves that the Cu and Fe nanomaterials formed in this current work are separate discrete nanomaterials and as such will be able to be separated from each other (e.g. selective dissolution). Physical and chemical properties of the Cu-bearing nanoparticles formed due to the exposure of 1000 mg/L solutions of CuCl2 and CuSO4 to 0.1 g/L nZVI for 4 h. It can be noted that both types of nanomaterial are polycrystalline with a relatively high sphericity. The nanoparticles formed from CuCl2 salts were typically of lower purity than those derived from CuSO4 salts, with the maximum Cu purity detected of 48.4 and 83.3 wt% Cu, respectively. The nanoparticles derived from CuSO4 salts were also recorded to be smaller than those derived from CuCl2 and also more rounded in shape.

TEM images of the nZVI after 4 h reaction with a 1000 mg/L CuCl2 solution, along with EDX maps of the four most common elements (by wt%) present. Composition (wt%) recorded using EDX for the overall map and spot (marked in red) is also displayed. An nZVI concentration of 0.1 g/L was used. TEM: transmission electron microscopy; EDX: energy dispersive X-ray; nZVI: nanoscale zerovalent iron.

TEM images of the nZVI after 4 h reaction with a 1000 mg/L CuSO4 solution, along with EDX maps of the four most common elements (by wt%) present. Composition (wt%) recorded using EDX for the overall map and spot (marked in red) is also displayed. An nZVI concentration of 0.1 g/L was used. TEM: transmission electron microscopy; EDX: energy dispersive X-ray; nZVI: nanoscale zerovalent iron.
Zeta potential data
Figure 12 displays zeta potential (ζ) measurements as a function of solution pH for the Cu-bearing nanoparticles which have formed from the exposure of the nZVI to CuCl2 or CuSO4 solutions at 1000 mg/L for 24 h. It can be seen that a different ζ range was recorded for the nZVI which have been exposed to CuCl2 or CuSO4 solutions, with maximum of 8.5 and 16.2 mV recorded for each nanoparticle type at a pH of 3.3 and 3.2, respectively, and a minimum of −14.0 and −16.2 mV recorded for each nanoparticle type at a pH of 12.1 and 11.9, respectively. The point of zero charge (PZC) is recorded as much lower for the nZVI which have been exposed to CuCl2 solution compared to the nZVI which have been exposed to the CuSO4 solutions, with a PZC recorded to be at a pH of approximately 6.7 and 10.5, respectively. The differences in ζ versus pH (along with the XRD data displayed in Figure 8) further confirm that the products formed in the CuSO4 and CuCl2 systems are physicochemically distinct. It is often assumed that colloids with a ζ of >40 mV possess high stability in the aqueous phase. 28 It can therefore be stated that both nanomaterial types are likely to exhibit relatively limited long-term stability in the aqueous phase.

Zeta potential (mV) as a function of pH for the nanoparticles formed following the exposure of nZVI (concentration of 0.1 g/L) to 1000 mg/L CuCl2 and CuSO4 solutions for 24 h. nZVI: nanoscale zerovalent iron.
Industrial/environmental implications
Electrowinning and electrorefining are the most common conventional routes for the recovery of Cu from both aqueous sources (waste water, lixiviants, etc.) and impure metals (e.g. blister Cu), respectively, with the objective of both methods the formation of a high-purity, often highly crystalline, Cu electroplate deposit at the cathode. The resultant Cu sheets are then extracted and sold directly as a raw material which are then subsequently transformed into Cu-bearing products for uses in a wide range of applications. The synthesis of Cu nanoparticles from such sheets is then typically conducted via: (i) dissolution of the sheet into an electrolyte which is followed by a nanoparticle recovery method (e.g. chemical precipitation or chemical reduction) or (ii) mechanical, thermal or sonochemical decomposition (e.g. ball milling, ultrasound). A key economic limit associated with this process, therefore, is that these processes require several expensive and energy intensive steps. Instead if Cu nanoparticles could be synthesised directly from the aqueous Cu source (e.g. Cu-bearing mining leachate, Cu-bearing waste water), then the process would be fundamentally more efficient. Here we have demonstrated that nZVI could be used as a highly effective reagent to selectively produce high-purity Cu nanoparticles (83.3% Cu, >99.9% Cu and O) directly from aqueous Cu sources via a one-pot process that does not require any electrical input. The use of nZVI is also particularly useful because it is selective for Cu nanoparticle formation from mixed aqueous systems, 29,30 The results demonstrate that the process is highly adaptable and can result in the formation of high purity Cu-bearing nanoparticles of various different physical structures and chemistry. For example, Table 2 displays an overview of the different physical and chemical properties of Cu-bearing nanoparticles formed via the exposure of aqueous Cu (as 1000 mg/L solutions of CuCl2 and CuSO4) with 0.1 g/L nZVI for 4 h. In contrast, the use of a more powerful chemical reducing agent (such as NaBH4) would exhibit less selectivity than nZVI and thus chemically reduce a mixture of present metals (e.g. Cu, Fe, Co, Ag) resulting in a nanomaterial of mixed chemical composition that likely has limited functionality. This work therefore provides a key first step in the emerging research field into the development of next generation methodologies for the selective and direct formation of engineered nanomaterials from aqueous sources. In particular, this research could be particularly applicable for the selective recovery of Cu from aqueous waste streams (e.g. acid mine drainage, industrial effluents), where the significantly greater economic gain generated by the direct formation of high-value engineered nanomaterials (rather than Cu plates from electrowinning) could be used to significantly offset the cost of such waste water treatment processes.
Physical and chemical properties of the Cu-bearing nanoparticles formed due to the exposure of 1000 mg/L solutions of CuCl2 and CuSO4 to 0.1 g/L nZVI for 4 h.

XRD: X-ray diffraction; TEM: transmission electron microscopy; EDX: energy dispersive X-ray; nZVI: nanoscale zerovalent iron.
Conclusions
Waste valorisation is a critical research field because it acts to both safeguard the environment but also protect essential material supply chains. A relatively unexplored yet extremely promising strand of this research is the development of methodologies for the direct formation of functional nanomaterials from waste because it has the potential to impart dramatic cost savings due to the substantially high economic value of certain nanomaterials when compared to their bulk scale counterpart.
Here the influence of different chemical factors (namely, solid–liquid ratio, starting pH, starting Cu concentration and anion type) on the cementation of aqueous Cu with nZVI has been studied. The work has been established to investigate the mechanisms and kinetics of Cu removal from solution but in particular the physicochemical composition of resultant Cu-bearing cemented (nano)particles. The following can be concluded: The nZVI exhibited exceptionally high Cu removal capacity (maximum removal recorded was 905.2 mg/g) due to the high purity and reactivity of the nZVI. Starting pH exhibited a relatively strong influence on the extent of Cu uptake with greatest Cu removal from solution and retention in the solid phase recorded for batch systems at a starting pH of 4.0. Type of anion(s) present in the Cu-bearing solution exhibited a clear influence on both the extent of Cu removal but also the type of Cu nanoparticulate products formed, with Cu2O, CuCl2 and Cu2(OH)3Cl determined as the most common nanoparticulates formed for the batch systems containing Cl− as the counterion, while Cu0 and Cu2O were the most common phases formed in systems containing Starting pH exhibited no discernible influence on the bulk chemical composition (phase) of Cu nanoparticles formed via cementation of CuSO4 solutions with nZVI, with a mixture of Cu0 and Cu2O recorded for all system studies. In contrast, starting pH exhibited a clear influence on the bulk chemical composition of Cu nanoparticles formed via cementation of CuCl2 solutions with nZVI, with Cu2O recorded as the main phase present for Cu formed from CuCl2 solutions with a starting pH of 3.0, compared to a mixture of Cu2Cl(OH)3 and CuCl2 recorded for solutions with a starting pH of 4.0. TEM analysis determined the Cu-bearing nanoparticles to be extremely high purity (e.g. >80 wt% Cu or ≥99.9 wt% Cu and O) which were recorded as among, but not chemically bound to, needle-shaped nZVI amorphous corrosion products. Solid liquid separation of Cu-bearing nanoparticles must occur relatively quickly after nZVI addition (e.g. within 4 h) to prevent Cu re-release (redissolution).
Overall the results demonstrate nZVI as a highly effective scavenger of aqueous Cu, with the one-pot formation of discrete high-purity Cu-bearing nanoparticles which could then be directly used in a wide range of applications. Future work will investigate how nZVI selectivity for metal high-purity nanoparticle formation can be fine-tuned for operation in increasingly complex environments (e.g. mixed contaminant systems and/or water bodies containing complex combinations of background solutes). In addition, the sourcing of nZVI from cheaper materials (e.g. waste streams) would also be highly beneficial to improve the economic return of such processes.
Footnotes
Acknowledgements
We would like to thank Mr Jeff Rowlands and Mr Marco Santonastaso and from the School of Engineering, Cardiff University for their technical support. We would also like to thank Dr Thomas Davies from the Cardiff Catalysis Institute and the Cardiff Electron Microscopy Facility for the TEM analysis. Photographs included in the RHS of the graphical abstract are from the following references (clockwise from top LHS): 31 –35
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Natural Environment Research Council (grant no. NE/L013908/1) and the Camborne School of Mines Trust.