
Abstract
Keywords
Introduction
Genetic conditions, caused partially or entirely by variations in the DNA sequence, can significantly affect the growth, development and health of individuals (National Human Genome Research Institute, 2015; O’Malley and Hutcheon, 2007). These conditions are broadly categorised as chromosomal, monogenic (single gene) and multifactorial (Nussbaum et al., 2015). Advances in genomic technologies, such as next-generation sequencing, have reshaped the understanding and classification of genetic conditions, facilitating improved diagnostics and the enhanced identification of genetic variants, including incidental findings and variants of uncertain significance (Mattick et al., 2014; Stark and Scott, 2023; Stranneheim and Wedell, 2016). The introduction of genomic newborn screening has significantly expanded the range of conditions detectable early in a child’s life, supporting earlier interventions (Downie et al., 2021, 2024; Lunke et al., 2024; Lynch et al., 2024). Similarly, developments in prenatal diagnostics, such as non-invasive prenatal testing and chromosomal microarray, have further enhanced the early detection of chromosomal anomalies, thereby contributing to increased notifications in congenital anomaly registers (Hui et al., 2017; MacArthur et al., 2023).
Despite diagnostic advancements, accurately estimating the prevalence and burden of genetic conditions remains complex. Reliable prevalence data are essential for effective public health planning, resource allocation and the development of targeted interventions aimed at reducing morbidity and mortality. Globally, around 6% of children are born with a serious congenital anomaly of genetic or partially genetic origin, contributing significantly to paediatric hospitalisations (Christianson et al., 2006). While Australia reports lower rates compared to some countries, congenital anomalies still account for a substantial proportion of perinatal deaths in Australia, with over 30% attributed to genetic or congenital defects (Australian Institute of Health and Welfare, 2024).
One major challenge in quantifying the burden of genetic conditions lies in ascertainment. That is, how cases are identified and recorded in official data. Globally, genetic conditions may be captured using various methods, including medical record reviews, International Classification of Diseases (ICD) coded administrative data, and clinical or disease-specific registries (Peng et al., 2018; Ruseckaite et al., 2023). Each method has distinct strengths and limitations. Medical record reviews provide detailed information but often use smaller samples, limiting generalisability (Dye et al., 2011; McCandless et al., 2004). ICD-coded administrative databases cover larger populations, but clinical coding practices may result in under-ascertainment or misclassification, particularly when genetic conditions are secondary diagnoses (Assareh et al., 2016; Riley et al., 2024). Clinical registries offer longitudinal tracking of specific conditions, yet their effectiveness varies due to differences in participation, scope and data-entry standards (Ruseckaite et al., 2023). Collectively, these differing methodologies and data sources can produce inconsistent prevalence figures, complicating efforts to monitor trends, plan services or compare results across regions.
Understanding disease frequency (prevalence and incidence) has significant implications. These measures not only inform the public health impact of genetic conditions but are crucial for targeted resource allocation, effective screening and the development of tailored preventive and therapeutic strategies (Abouelhoda et al., 2016; Colburn and Lapidus, 2024). Given these implications, a clear understanding of how genetic conditions are ascertained, and how their prevalence is reported, is paramount.
Aims
The purpose of this scoping review was to determine how different data sources and ascertainment methods influence the reported prevalence of paediatric monogenic and chromosomal conditions in Australia, New Zealand, Europe and North America. Specifically, the review aimed to:
Identify the key data sources used to capture monogenic and chromosomal conditions.
Examine how varying ascertainment methods shape the reported prevalence of these conditions.
Describe how disease prevalence is presented in the peer-reviewed literature.
Highlight gaps in ascertainment and notification practices and propose areas for improvement in Australia relative to international findings.
Method
Study design
A scoping review of the literature was conducted using the Arksey and O’Malley (2005) framework and further supplemented by the Joanna Briggs Institute guidelines (Peters et al., 2020) and a modified Preferred Reporting and Meta-Analysis extension for scoping reviews checklist (Tricco et al., 2018). A protocol (non-registered) outlining the objectives, inclusion criteria and planned methods was developed a priori.
Search strategy
To address the research objectives, a structured population, context and concept search strategy (Peters et al., 2020) was conducted in consultation with a specialist librarian, to systematically identify relevant peer-reviewed literature (Box 1). During January 2025, an independent search of Medline, CINAHL and SCOPUS electronic databases was undertaken by one author (SG) using the agreed search terms. These databases were chosen for their coverage of biomedical, epidemiological and health services research. Google Scholar was also utilised; however, due to the high number of results, only the first 200 entries were considered. Reference lists of literature which met the inclusion criteria were manually searched (SG) to identify additional relevant publications. Titles that included the following terms were assessed for suitability for inclusion; “prevalence,” “frequency,” or “burden.”
Key search terms.

Used to retrieve variations on a distinctive word stem or root (e.g. chromosom* = chromosome and/or chromosomal).
Selection of genetic conditions
The foci of this scoping review were chromosomal and monogenic conditions (Box 2), rather than all genetic conditions, as advances in disease knowledge have blurred the distinction between genetic and non-genetic conditions (Dye et al., 2011). Multifactorial conditions fall outside the scope of the review, as they involve non-Mendelian inheritance, with traits influenced by both genetic and environmental factors (Nussbaum et al., 2015). To confirm that conditions met the criteria, inheritance patterns were verified using Online Mendelian Inheritance in Man (OMIM, 2025), Orphanet (2024a) and supplementary material from Gjorgioski et al. (2020). OMIM (2025) is a continuously updated, comprehensive database of human genes and genetic phenotypes, focusing particularly on the relationship between phenotype and genotype. Orphanet (2024a) is a European-based reference portal that provides detailed information on rare diseases, including their clinical presentation, inheritance patterns and classification.
Classification and definition of monogenic and chromosomal conditions.

Eligibility criteria
Peer-reviewed articles published between 2004 and 2024, inclusive, were considered to ensure a focus on the relevant contemporary literature. No language restrictions were applied initially; however, only studies available in English proceeded to full-text screening. To be eligible for inclusion, studies needed to report on at least one monogenic or chromosomal condition. This could be through individual analysis, grouping under categories such as “monogenic” or “chromosomal,” or as part of broader research on rare diseases or birth defects, even if not all conditions met strict genetic definitions. Studies that did not clearly distinguish genetic conditions from broader birth defects (regardless of genetic contribution) were excluded from the review. Furthermore, studies had to include data on children aged under 6 years, as many genetic and congenital conditions are diagnosed beyond infancy and emerge during early childhood (Bower et al., 2010; Gibson et al., 2016). Studies involving broader age groups, including older children or adults, were included only if they reported extractable data specific to children under 6. This criterion ensured the review focused on prevalence and ascertainment during the early years of life. To maximise the comparability and applicability of findings to the Australian context, only studies from New Zealand, Europe and North America were included. These regions share socio-economic conditions, healthcare systems, and demographic characteristics sufficiently similar to Australia (Organisation for Economic Co-operation and Development, 2019) to enhance the relevance and transferability of the review’s conclusions.
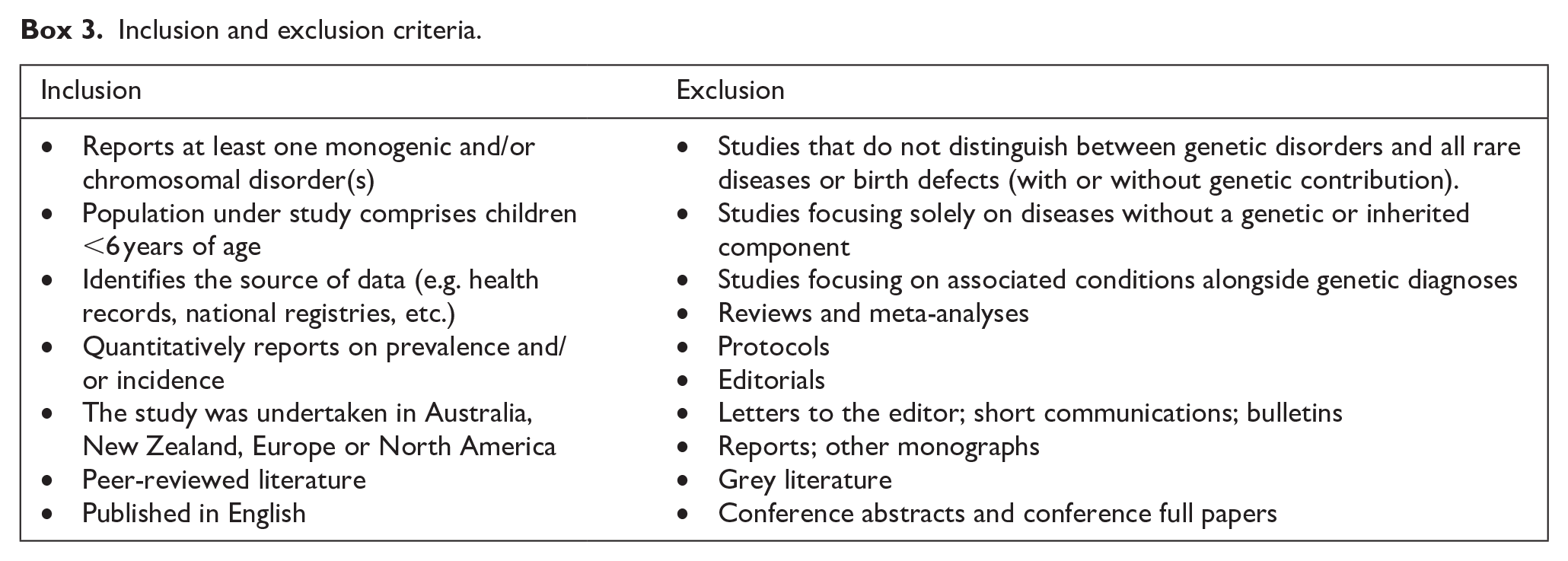
In addition, research focusing on specific patient groups or subpopulations were excluded, as these do not reflect general population prevalence. For instance, a study examining obstructive sleep apnoea in individuals with Down syndrome was not included because it did not represent the overall prevalence of the genetic condition itself. Finally, to align with the study’s objectives, articles were required to report prevalence or frequency estimates. Studies not meeting these criteria were excluded. The screening criteria are summarised in Box 3.
Inclusion and exclusion criteria.
Eligibility screening
Eligibility screening was conducted in two stages using the online reference management tool, Covidence Systematic Review Software (2024). First, two reviewers (SG, MT) independently assessed titles and abstracts against the inclusion criteria. Discrepancies were resolved by a third reviewer (MR). All records not excluded were passed on for full-text review. Two reviewers (SG, MR) independently reviewed full-text records for potentially eligible studies and any disagreements were resolved via discussion (SG, MR). Records deemed ineligible at full-text screening were excluded with the reason recorded.
Data extraction
Following full-text review, data extraction was undertaken by three reviewers (SG, MR and MT) using Covidence Systematic Review Software (2024). Where there was uncertainty about what information to extract, a second author reviewed the article, and discrepancies were resolved through discussion among the reviewers. A data extraction form was developed and pilot-tested on a subset of articles to ensure clarity and comprehensiveness. To minimise bias, data extraction for the article authored by members of the review team (SG, MR) was conducted by an independent reviewer (MT) who was not involved in its authorship. The following bibliometric and research details were extracted from all eligible studies: study identification number; title; name of lead author; (first) year of publication (online or journal issue); title of journal; country; geographical region; study aims; study design; study setting; study time period; participants’ age; sample size; genetic condition(s); genetic inheritance type; source of data (i.e. hospital records, administrative dataset, disease registry, etc.); name of the data source; case ascertainment (active/passive); case ascertainment methods (i.e. medical record abstraction, ICD coding system, genetic testing); how prevalence is reported and prevalence data.
In this review, surveillance approaches were categorised as either active or passive, depending on how data collection was conducted. Active surveillance refers to a process where personnel systematically contact healthcare providers or individuals to obtain information about health conditions. This approach yields highly accurate and timely data but is resource-intensive and costly (Nsubuga et al., 2006). Conversely, passive surveillance involves the receipt of health data from external sources such as hospitals, clinics or public health units. While more economical and capable of covering large populations, passive systems may suffer from variability in data quality and timeliness due to reliance on voluntary reporting (Nsubuga et al., 2006).
Data analysis
Descriptive analyses, supported by Microsoft Excel (Microsoft, 2025), were utilised to summarise the results. Frequencies and percentages were calculated to present the distribution of studies by year of publication, geographic region, genetic condition type, data sources and ascertainment methods. Articles that focused on more than one condition and could be classified as either chromosomal or monogenic were grouped into a “Complex and Mixed Genetic Condition” category.
Ethical approval
Ethics approval was not required because this scoping review did not involve animal or human participants.
Results
Selection of articles
The search of Medline, CINAHL, Scopus and Google Scholar yielded a total of 2429 articles (Figure 1). A further 20 articles were identified through manual reference review of eligible studies. After removing duplicates, 1894 abstracts were reviewed, and 302 full-text articles were assessed for eligibility. A total of 58 articles met the inclusion criteria.
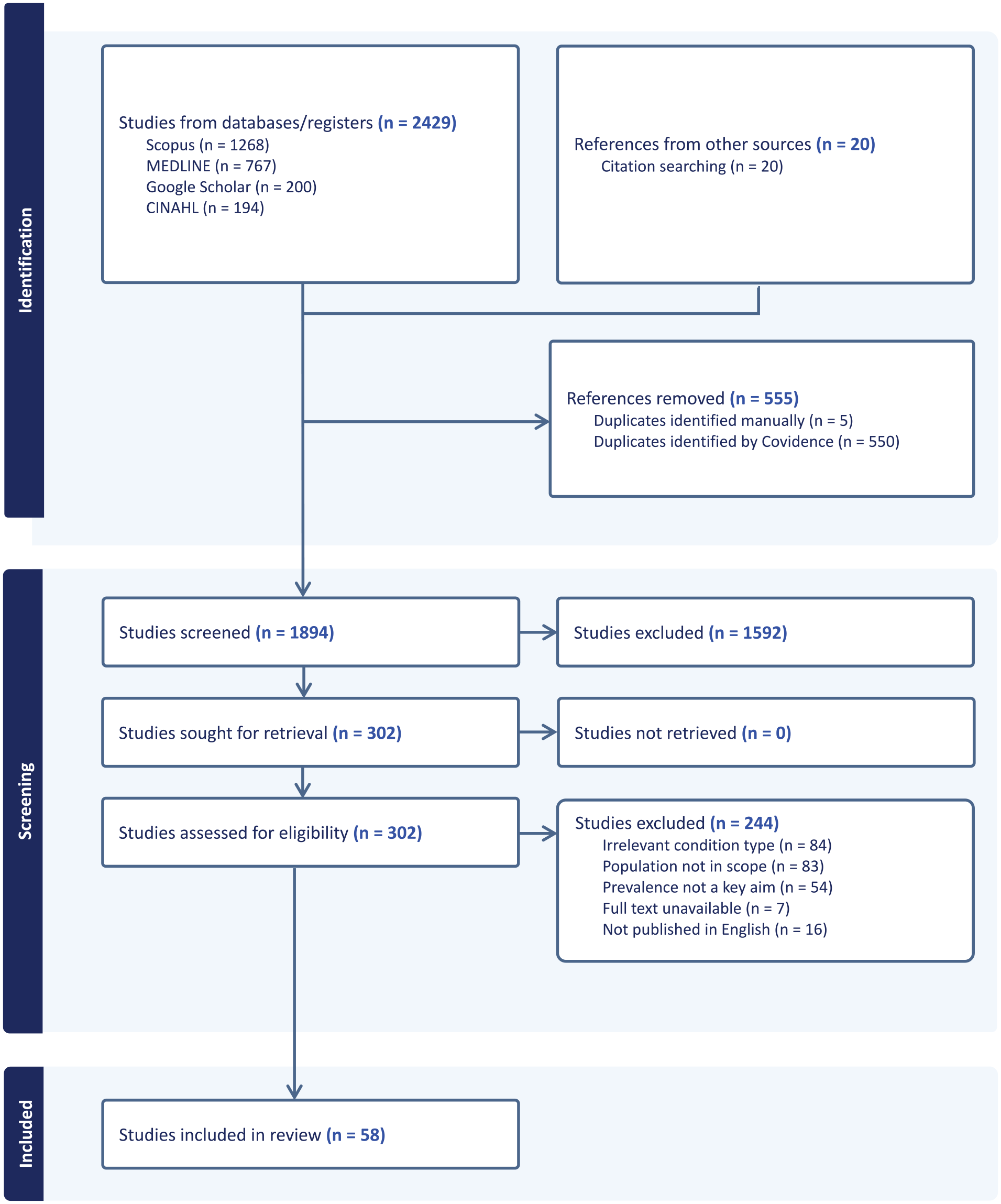
Preferred Reporting Items in Systematic Reviews and Meta-Analysis (PRISMA) flowchart of scoping review (Covidence generated).
Characteristics of studies
Table 1 summarises the key characteristics of the within-scope articles. Most of the articles (77.6%) were published from 2010 onwards; with 34.5% (n = 20) published between 2019 and 2024. Based on the source of the data, most publications originated from Europe (58.6%, n = 34), followed by North America (31.0%, n = 18) and Australia (5.2%, n = 3). There were no studies originating from New Zealand. Within Europe, four studies focused on England and Wales and three on Denmark, whereas seven spanned multiple European countries. In North America, the United States of America (USA) accounted for the majority (17 articles, or 29.5% of all included studies) and also represented the highest number of publications from a single country. A smaller fraction (5.2%) of articles involved data or collaborations across multiple regions (e.g. United Kingdom (UK) and Australia; Europe and Israel).
Summary of key characteristics of studies reported in within-scope articles.
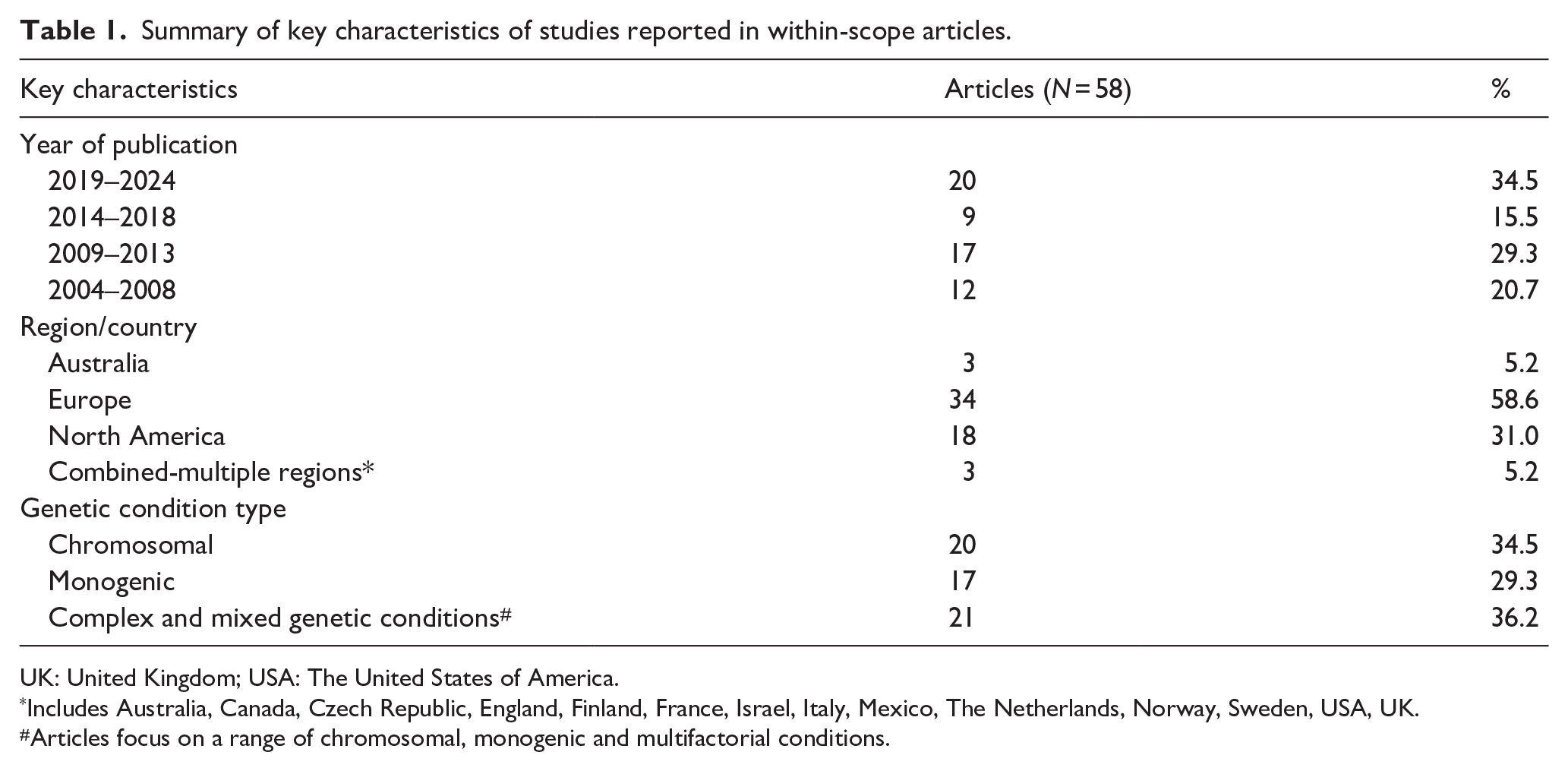
UK: United Kingdom; USA: The United States of America.
Includes Australia, Canada, Czech Republic, England, Finland, France, Israel, Italy, Mexico, The Netherlands, Norway, Sweden, USA, UK.
Articles focus on a range of chromosomal, monogenic and multifactorial conditions.
Types of genetic conditions
Of the 58 included articles, 34.5% (n = 20) focused on chromosomal conditions, 29.3% (n = 17) on monogenic conditions and 36.2% (n = 21) on complex or mixed genetic conditions (Table 1). Among the chromosomal disorder studies, more than half (n = 11) of the reported studies investigated Down Syndrome exclusively. Articles categorised under monogenic, examined conditions such as X-linked agammaglobulinemia and Mucopolysaccharidosis type 1. Studies grouped into the complex and mixed genetic condition category encompassed a broad range of chromosomal, monogenic and multifactorial conditions. Key examples of data that were extracted include Trisomy 21, 18 and 13, as well as more common single-gene disorders such as Cystic Fibrosis, Thalassemia and Osteogenesis Imperfecta.
Sources of data
Data sources used to assess the prevalence of monogenic and chromosomal conditions in the included studies highlighted a strong reliance on disease registries; these constituted the most utilised source, appearing in 62.1% (n = 41; Table 2). Among the most frequently used registries were the European Registration of Congenital Anomalies and Twins (EUROCAT; n = 10), a European network for monitoring congenital anomalies and the National Birth Defects Prevalence Network (NBDPN; n = 4), which collects and analyses data on birth defects in the USA. Hospital and health service records accounted for 19.7% (n = 12), while population-based administrative datasets were used in 12.3% (n = 8) of the reported studies. A smaller proportion of studies (6.2%, n = 4) relied on other sources, including clinical laboratories, vital statistics and newborn screening programs. In the three Australian studies data sources included hospital and health service records (n = 1), population-based administrative datasets (n = 2) and clinical laboratories (n = 1). Conversely, only one study using Australian data utilised a disease registry; however, this was part of an international collaboration with researchers from the UK. Since authors of the articles could use more than one data source, some studies incorporated multiple sources (n = 8), combining, for example, disease registries with administrative datasets. Most studies (n = 50), however, relied upon a single source of data, with disease registries being the sole source in 36 articles, followed by hospital and health service records (n = 10) and population-based administrative datasets (n = 3).
Sources of data used for case ascertainment.
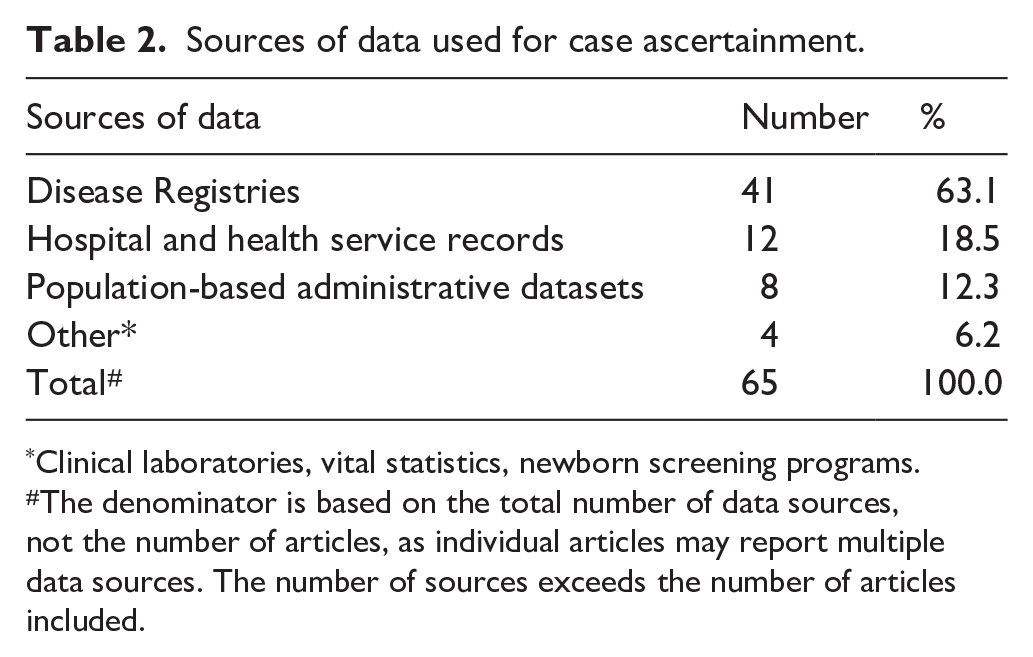
Clinical laboratories, vital statistics, newborn screening programs.
The denominator is based on the total number of data sources, not the number of articles, as individual articles may report multiple data sources. The number of sources exceeds the number of articles included.
Case ascertainment methods
Case ascertainment methods varied across studies, with the majority employing active ascertainment (77.6%, n = 45). Notably, active case ascertainment was used in most studies that relied upon disease registries, perhaps reflecting the more structured and systematic approach that these registries employ for data collection. Passive ascertainment was less common (6.9%, n = 4) and was predominantly used in studies relying on population-based administrative datasets. A mixed approach combining both active and passive methods was used in 13.8% (n = 8).
In implementing these ascertainment strategies, authors of studies used various types of case identification methods, often applying more than one method to improve case ascertainment (Table 3). Medical record abstraction was the most frequently used method (29.7%, n = 27), followed closely by ICD and other coded data (27.5%, n = 25) and genetic testing with laboratory confirmation (27.5%, n = 25). Health service or clinician-reported cases (8.8%, n = 8) and registry linkage (4.4%, n = 4) were used less frequently. While 28 studies relied on a single ascertainment method, a substantial proportion (n = 30) incorporated multiple sources, such as combining genetic test results with administrative data or medical record review. The three Australian studies demonstrated variability in how genetic conditions were ascertained. Dye et al. (2011) relied on passive ascertainment through ICD-coded data, whilst Gjorgioski et al. (2020) and Hui et al. (2020) employed active ascertainment methods incorporating medical record abstraction, genetic testing and laboratory confirmation.
Methods of case ascertainment.
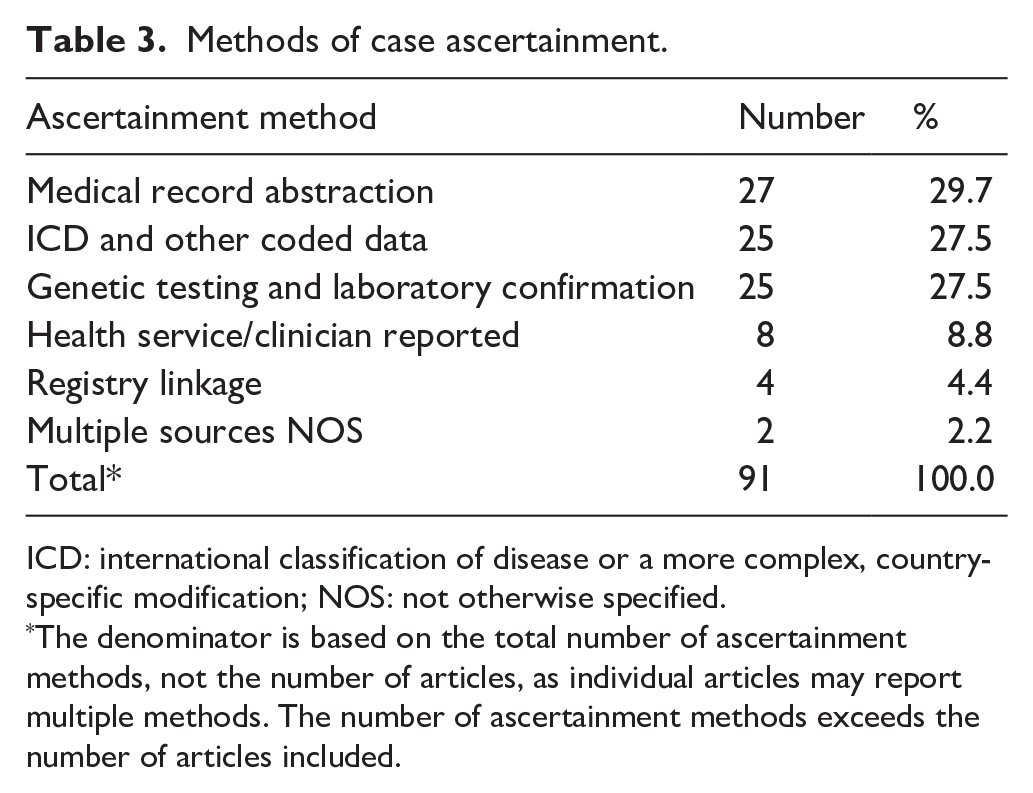
ICD: international classification of disease or a more complex, country-specific modification; NOS: not otherwise specified.
The denominator is based on the total number of ascertainment methods, not the number of articles, as individual articles may report multiple methods. The number of ascertainment methods exceeds the number of articles included.
Studies that utilised coded data, mostly used ICD-based coding systems (ICD-10 and ICD-9 versions), including the respective clinical modifications (CMs) and the Australian modification (AM). The British Paediatric Association (BPA) extension, used to code congenital anomalies, was frequently mentioned alongside ICD-9 and ICD-10. Additionally, some studies incorporated genetic condition-specific databases such as OMIM and Orphanet, often alongside ICD-coded data.
Prevalence of chromosomal and monogenic conditions as reported in the literature
The reporting of prevalence varied across studies, reflecting differences in data sources, study populations, and case ascertainment methods. The sample size reported varied, ranging from 31 to 12,886,464 participants/admissions. Studies based on large-scale administrative datasets or national registries tended to have substantially larger sample sizes than those relying on single disease registries.
Prevalence estimates were commonly expressed in relation to population size, most frequently per number of births or per total population (Table 4). Per number of births was the most frequent approach (n = 22) reflected in studies using disease registries, particularly those focusing on congenital or early-onset genetic conditions. For example, Fraser Syndrome was reported at 0.2 per 100,000 births (Barisic et al., 2013), while Holt-Oram Syndrome had a prevalence of 0.7 per 100,000 births (Barisic et al., 2014), both derived from EUROCAT. Similarly, studies examining Down Syndrome reported prevalence of between 11.8 and 22.0 per 10,000 births using disease registries (Loane et al., 2013; Shin et al., 2009), and 7.01 per 10,000 using population administrative datasets (Glivetic et al., 2015).
General description and key characteristics of studies identified in the scoping review.
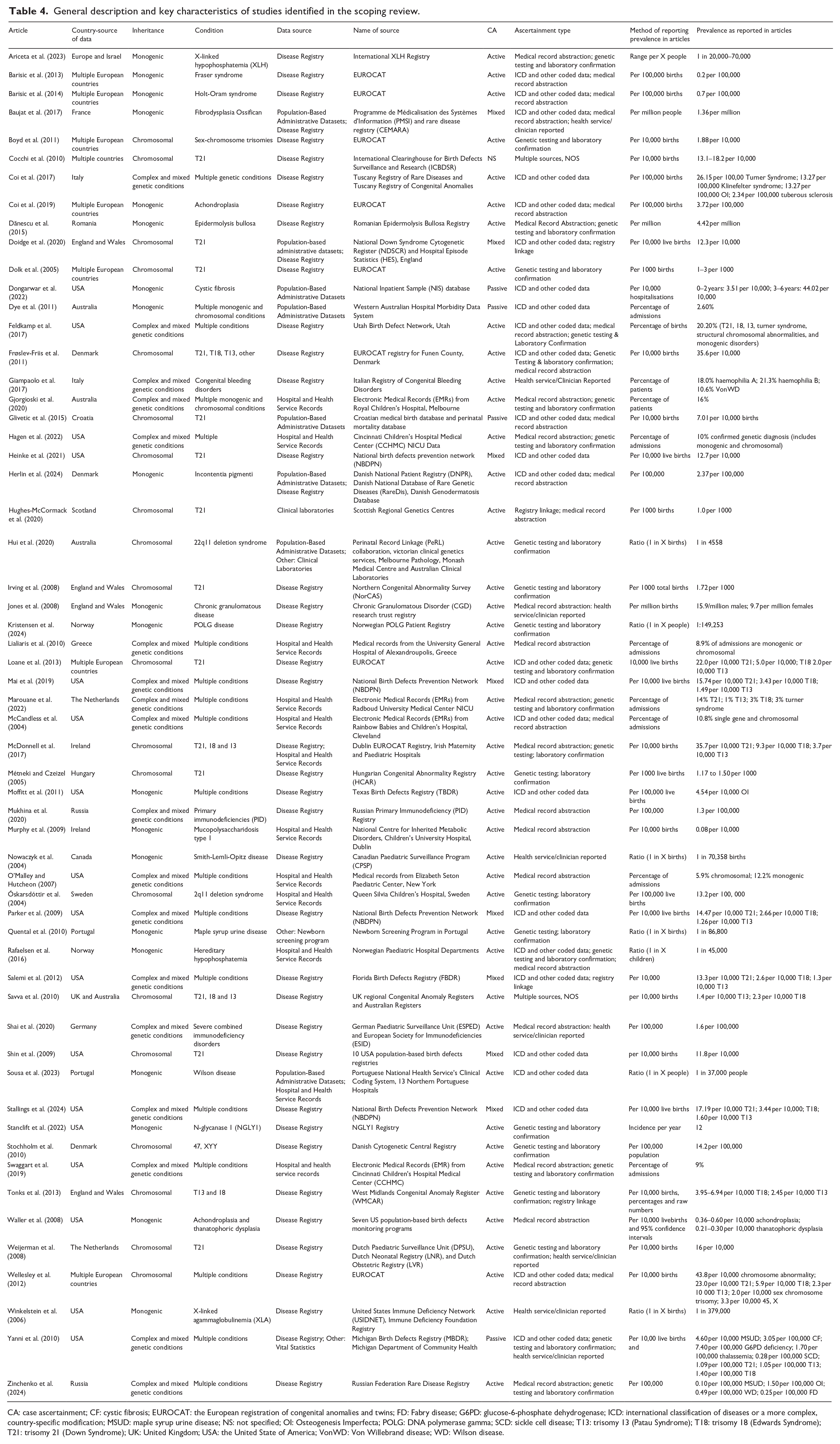
CA: case ascertainment; CF: cystic fibrosis; EUROCAT: the European registration of congenital anomalies and twins; FD: Fabry disease; G6PD: glucose-6-phosphate dehydrogenase; ICD: international classification of diseases or a more complex, country-specific modification; MSUD: maple syrup urine disease; NS: not specified; OI: Osteogenesis Imperfecta; POLG: DNA polymerase gamma; SCD: sickle cell disease; T13: trisomy 13 (Patau Syndrome); T18: trisomy 18 (Edwards Syndrome); T21: trisomy 21 (Down Syndrome); UK: United Kingdom; USA: the United State of America; VonWD: Von Willebrand disease; WD: Wilson disease.
Correspondingly, some studies expressed prevalence to the general population, particularly for conditions that impact individuals across the lifespan. For example, Fibrodysplasia Ossificans Progressive was reported at 1.36 per million people, using data from a population-based administrative dataset and disease registry in France (Baujat et al., 2017). While Epidermolysis Bullosa was reported at 4.42 per million people using a disease registry in Romania (Dănescu et al., 2015). Moreover, in some studies, the prevalence was reported using ratio-based estimates expressing the frequency of genetic conditions as 1 in X births, children or people. For instance, Nowaczyk et al. (2004), using data from the Canadian Paediatric Surveillance Program, estimated the prevalence of Smith-Lemli-Opitz Disease as 1 in 70,358 births, whilst Hui et al. (2020) estimated the prevalence of 22q1 Deletion Syndrome as 1 in 4558 births in Victoria, Australia (excluding miscarriages but including stillbirths and terminations), reflecting differences in study populations and ascertainment methods.
Prevalence estimates were also reported at times using all pregnancy outcomes rather than live births alone. Heinke et al. (2021) reported a Down Syndrome prevalence of 12.7 per 10,000 live births, increasing to 13.3 per 10,000 when terminations and stillbirths were included from the USA. Similarly, Cocchi et al. (2010) found that the total prevalence of Down Syndrome from 14 participating countries, including live births, stillbirths and terminations, increased from 13.1 to 18.2 per 10,000 births between 1993 and 2004. In contrast, the prevalence of live births alone remained stable at 8.3 per 10,000. This increase in total prevalence was accompanied by a rise in terminations of pregnancy for Down Syndrome, from 4.8 to 9.9 per 10,000 births over the same period.
Eleven studies reported the prevalence of conditions as a percentage of admissions or patients. The majority of these studies (n = 7/10) utilised health or hospital records, primarily through medical record abstraction. Studies conducted in Greece and the USA examining the impact of monogenic and chromosomal conditions collectively via medical record review found that these conditions accounted for 8.9%–18.1% of admissions (Lialiaris et al., 2010; McCandless et al., 2004; O’Malley and Hutcheon, 2007). In Australia, Gjorgioski et al. (2020) reported a 16% prevalence of admissions for these conditions in a Victorian hospital using a similar approach. In contrast, Dye et al. (2011), who analysed ICD-10-AM coded data from a population-based administrative dataset in Western Australia, found a substantially lower prevalence of 2.6% of admissions.
Discussion
This scoping review explored the data sources used to identify and record monogenic and chromosomal conditions in Australia, New Zealand, Europe and North America, as reported in the peer-reviewed literature between 2004 and 2024, inclusive. The review examined how different data sources influence prevalence estimates, and highlighted variations in case ascertainment and notification systems. Nearly 60% of the within-scope studies originated from European countries and 30% from the USA, illustrating the concentration of genetic disease research in these regions. In contrast, Australian authors accounted for only four studies, one of which involved international collaboration with researchers from the UK. The dominance of European and USA-based studies is likely driven by well-established surveillance networks and large-scale registries, such as EUROCAT, the NBDPN and the Genetic and Rare Diseases Information Centre. Additionally, Orphanet plays a key role in European rare disease surveillance, offering a structured and standardised framework for data collection across multiple countries (Orphanet, 2024a). Central to this system is the use of ORPHAcodes, which are unique, stable identifiers assigned to rare diseases within the Orphanet nomenclature.
Widely recognised as the most appropriate coding system for rare diseases, ORPHAcodes offer comprehensive coverage across Europe and are increasingly being integrated into broader classification systems, such as the ICD (Orphanet, 2024b; Rare Disease Awareness Rare Portal, 2024). These centralised systems facilitate consistent case ascertainment, national and cross-national level prevalence reporting and robust data linkage across healthcare systems and settings (Bascom et al., 2023; Dolk, 2005).
Australia, by comparison, does not have a centralised national genetic or rare disease registry. Instead, surveillance relies on a fragmented system of specialised registries, including the Australian Congenital Anomalies Monitoring System, the Australian Paediatric Surveillance Unit Database and state-based registries such as the Victorian Congenital Anomalies Register. Ruseckaite et al. (2023) highlight the variability in Australian rare disease registries in terms of data entry, scope, outputs and funding, and associated challenges of interoperability. There exist Australian disease-specific registries for some conditions, such as the Australian Cystic Fibrosis Data Registry and the Australian Rett Syndrome Database; however, many genetic conditions (particularly monogenic and chromosomal) lack dedicated national registries. This arrangement contributes to fragmented data, underrepresentation in surveillance and difficulties in estimating true prevalence (Elliot et al., 2024). In their scoping review, Ruseckaite et al. (2023) found only 24 Australian-only rare disease registries and five Australian jurisdiction-based registries. Given that over 7000 rare genetic conditions have been identified, around one-third of which affect children (Lee et al., 2020), a more coordinated effort is required to ensure comprehensive data collection (Rare Voices Australia and Monash University, 2023; Ruseckaite et al., 2023) to achieve more accurate prevalence estimates.
The current review identified considerable heterogeneity in data sources, case ascertainment methods and approaches to reporting prevalence for monogenic and chromosomal conditions. The reliance on disease registries as the primary data source for determining prevalence, evident in 62.1% of the within-scope studies, underscores their recognised role in systematically capturing rare conditions (Hageman et al., 2023; Ruseckaite et al., 2023). High usage of registries such as EUROCAT and the NBDPN reflects their extensive reach, standardised data collection protocols and longstanding contributions to congenital anomaly surveillance (Dolk, 2005; Mai et al., 2019). Nevertheless, heavy dependence on a single registry can introduce limitations, including variations in case definitions, incomplete coverage and under-ascertainment of milder or late-onset conditions (Nassar et al., 2007). Despite the widespread use of disease registries internationally, only one within-scope study incorporated disease-specific registry data from Australia (Savva et al., 2010), highlighting the limited integration of local registry data in prevalence estimates. Notably, over three-quarters of the within-scope studies relied upon a single data source, thereby limiting opportunities for data triangulation (Rutherford et al., 2010) and consistency checks (Molster et al., 2012). Notwithstanding, or perhaps, in recognition of these challenges, a substantial proportion of studies (n = 30) adopted multiple ascertainment methods to enhance data robustness, such as combining genetic testing with administrative data or medical record review. This mixed approach facilitates improved sensitivity and specificity in case identification (Saczynski et al., 2013).
Case ascertainment methods also varied across studies, with active ascertainment being the predominant approach (77.6%); aligned with the high proportion of studies using disease registries which employ structured and systematic methods for case identification (Boyle et al., 2018). Active ascertainment, including medical record abstraction (29.7%) and genetic testing with laboratory confirmation (27.5%), improves case completeness and diagnostic accuracy, particularly for conditions requiring molecular confirmation (Molster et al., 2016). In contrast, passive ascertainment was less common (6.9%) and typically linked to population-based administrative datasets that rely on existing coded data rather than proactive case identification, which can result in underreporting (Reichard et al., 2016).
These methodological differences contribute to variations in prevalence estimates, as studies using disease registries from Europe and the USA, tend to report higher disease estimates due to more comprehensive case capture and larger sample sizes. For example, the reported prevalence of Down Syndrome ranged from 4.79 to 14.99 per 10,000, depending on whether population administrative datasets or disease registries were used. This discrepancy is likely because disease registries actively identify and verify cases through multiple data sources. Administrative datasets may miss cases that are asymptomatic, not coded as a primary diagnosis or diagnosed outside hospital settings. Moreover, administrative datasets in Australia primarily use ICD coded for disease surveillance, which can lead to underestimation (Liu et al., 2022; Ryan et al., 2021). These findings underscore the importance of integrating multiple data sources to improve the accuracy and reliability of genetic disease surveillance.
Furthermore, prevalence estimates of genetic conditions can differ notably based on whether only live births or all pregnancy outcomes (including terminations and stillbirths) are considered. For example, Heinke et al. (2021) reported an increase in the prevalence of Down Syndrome when terminated pregnancies and stillbirths were included. This discrepancy underscores how the inclusion of non-live-birth outcomes can substantially influence reported prevalence, particularly for conditions frequently detected through prenatal screening (Hui et al., 2016; Hui and Halliday, 2023). Moreover, the extent of this variation may depend on factors such as maternal age and access to genetic testing (Hui et al., 2016). Additionally, comparing prevalence estimates across studies is further complicated by differences in denominator definitions. For example, where prevalence is reported per 10,000 or per 100,000 births, or as a percentage of patients. This inconsistency can obscure direct comparisons and limit the interpretability of findings, particularly when determining prevalence estimates for Australia. Consequently, when interpreting and comparing prevalence rates, it is essential to account for both the population denominator and whether all pregnancy outcomes are included.
Studies employing medical record abstraction offered a more detailed and nuanced approach to identifying genetic conditions by reviewing full patient records rather than relying solely on coded discharge diagnoses. Approximately half of these studies were conducted within hospitals and health services (n = 10) while the rest were based in disease registries. This method can uncover underlying genetic conditions that are not explicitly recorded via the coded data, resulting in more complete case ascertainment and improved disease estimates. For example, McCandless et al. (2004) and Gjorgioski et al. (2020) identified genetic components missed in ICD-coded data (Dye et al., 2011), showing that administrative datasets underestimate the burden of monogenic and chromosomal diseases. McCandless et al. (2004) reported that 25% of genetic conditions were not captured in coded data, a pattern echoed in Gjorgioski (2017, unpublished thesis) and other validation studies across different diseases such as stroke (Ryan et al., 2021).
In contrast, disease registries provide a systematic approach to genetic disease surveillance through active ascertainment, ensuring that diagnosed cases are recorded and followed over time. Registries often use standardised data collection protocols, improving consistency in case reporting. Unlike medical record reviews, however, registries are usually limited to specific diseases and may not capture undiagnosed or incidental cases, particularly if they rely on voluntary reporting or do not integrate genetic testing results.
Despite their advantages, medical record reviews are resource-intensive, typically confined to single health services and have smaller sample sizes compared to registries and administrative datasets. Their accuracy also depends heavily on clinician documentation. Issues such as copy-pasting of clinical notes, outdated patient histories, or missing updates to genetic diagnoses in electronic medical records (EMRs) can introduce biases (Al Bahrani and Medhi, 2023; Casey et al., 2016). These factors may lead to over- or under-estimation of prevalence, depending on how genetic information is recorded and updated.
While ICD-based coding systems were widely used (27.5%), including ICD-9 and ICD-10 (with country-specific CMs such as the ICD-10-AM), coding-based approaches may be subject to misclassification or incomplete case capture, especially for conditions with complex diagnostic criteria (Southern et al., 2016). The incorporation of the BPA extension for congenital anomalies, alongside rare disease-specific databases such as OMIM and Orphanet, emphasises the need for coding frameworks that can handle the wide range of genetic conditions (Hageman et al., 2023; Walker et al., 2017).
Nevertheless, Australia’s reliance on ICD-10-AM for coded health data poses significant challenges for the accurate capture of genetic conditions. Historically, many genetic conditions lack specific codes or are categorised under broad groupings (Bowker and Star, 1999), leading to underreporting and poor surveillance. For instance, while Cystic Fibrosis (E84) has a distinct classification, Long QT Syndrome (I49.8) is included under “Other specific cardiac arrhythmias,” in the ICD, thus limiting detailed monitoring. Likewise, a patient with Osteogenesis Imperfecta who is admitted for a broken wrist may have only the fracture coded and not the underlying genetic diagnosis (Lujic et al., 2014). In Australia, co-morbid conditions are coded only if they meet Australian Coding Standard 0002, which stipulates that the condition must impact upon patient care by requiring therapeutic treatment, diagnostic procedures or increased nursing care and/or monitoring (Independent Hospital Pricing Authority, 2019). Thus, genetic diagnoses that do not affect the current admission, need not be coded (Assareh et al., 2016; Lujic et al., 2014).
In 2015, supplementary codes were introduced in the 9th edition of the ICD-10-AM to capture chronic conditions present on admission but not actively treated during an episode of care (Lujic et al., 2017). These codes, however, are not comprehensive and do not sufficiently capture the burden of many genetic conditions, which perpetuate their underrepresentation in administrative datasets. The increasing use of genomic testing and the multisystemic nature of many genetic conditions (Mellis et al., 2022) further complicate their classification within a statistical coding framework. Australia’s National Strategic Action Plan for Rare Diseases notes that only 517 of the nearly 7000 Orphanet-recognised rare diseases can be coded in Australian hospitals (Department of Health, 2020). While Action 3.1.1 proposes the integration of ORPHAcodes and ICD-11, implementation remains uncertain because ICD-11 adoption is still several years away in Australia. The absence of a clear pathway for incorporating ORPHAcodes into the existing ICD-10-AM framework leaves a gap in the immediate integration of genetic disease data. Consequently, until the ICD-11 is adopted, a structured approach to addressing limitations in the Australian clinical coding system remains elusive, thus creating ongoing barriers to accurate genetic disease surveillance, research and policy development.
Strengthening clinical coder and Health Information Manager (HIM) expertise is equally critical, given the complexity of genetic diagnoses and diverse inheritance patterns. In the future, HIMs will likely be responsible for assigning ORPHAcodes at the hospital level, thereby contributing directly to national genetic disease estimates. As the frontline professionals responsible for the quality and accuracy of clinically coded health data, HIMs must be equipped with the knowledge and training necessary to ensure that genetic conditions are appropriately classified and captured within national health datasets. Targeted education in genetic nomenclature, inheritance mechanisms, and coding frameworks can empower HIMs and clinical coders to accurately identify, document, and classify genetic conditions to ensure the comprehensive capture of relevant information.
Strengths and limitations
This study has several strengths. These include its international scope and comparison of like systems, which have allowed for a broader understanding of how different countries capture monogenic and chromosomal disorders. Unlike many studies that focus on all genetic conditions or rare diseases (some of which are non-genetic), this review has specifically examined monogenic and chromosomal disorders, thereby providing a more targeted analysis of prevalence estimation challenges. The study limitations include its exclusion of grey literature and white papers which may have led to the omission of relevant reports containing prevalence data. Additionally, as this was a scoping review rather than a systematic review, there is a possibility that some studies were missed. Another potential limitation is the risk of misclassifying disease inheritance, which could have led to the incorrect inclusion or exclusion of certain conditions, although this was mitigated by using established genetic databases such as OMIM and Orphanet to guide classification. Although data extraction was performed by three independent reviewers, no formal statistical analysis or audit was conducted to evaluate inter-rater reliability or the consistency of data extraction. This may potentially introduce variability or extraction bias into the results. Despite these limitations, the study provides valuable insights into the complexities of disease surveillance and data collection for monogenic and chromosomal conditions.
Conclusion
The findings highlight the significant variability in the methods used to ascertain cases and report prevalence of monogenic and chromosomal conditions. This variability underscores the need for a multipronged approach to case ascertainment that integrates registry data, ICD-coded administrative data, and rare disease-specific classification systems such as ORPHAcodes. Such an approach would enhance the accuracy, consistency and comparability of prevalence estimates across settings. In Australia, addressing the fragmented surveillance infrastructure is essential to ensure the reliability of prevalence data for genetic conditions. Key opportunities include developing multi-jurisdictional registries to support uniform data collection nationwide and leveraging Systematized Nomenclature of Medicine -Clinical Terms (SNOMED-CT) within EMRs to better capture genetic phenotypes (Mellis et al., 2022). While these priorities are outlined in the National Strategic Action Plan for Rare Diseases (Department of Health, 2020), progress (particularly in relation to ORPHAcodes) remains limited. Achieving a more integrated surveillance system will require coordinated national leadership, sustained funding and investment in workforce capabilities. Strengthening these foundations will support the generation of robust, policy-relevant prevalence data for genetic conditions in Australia.
Footnotes
Acknowledgements
The authors thank Hannah Buttery, Librarian at La Trobe University Library, Melbourne, for expert advice during the database searches.
Accepted for publication June 10, 2025.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.