
Abstract
Background:
Pulmonary hypertension associated with systemic sclerosis (SSc-PH) carries a significant risk of mortality. Although interstitial lung disease (ILD) often coexists with SSc-PH, its impact on prognosis varies considerably depending on disease extension. The prognostic implications of different degrees of functional impairment secondary to ILD in patients with SSc-PH remain poorly understood.
Objectives:
This study aimed to stratify patients with SSc-PH according to their functional parameters and to identify potential prognostic factors in different patterns of ILD extension.
Design:
A retrospective study was conducted on consecutive patients with SSc-PH diagnosed according to the European Society of Cardiology/European Respiratory Society guidelines for PH since 2015, from two Italian Scleroderma Centers.
Methods:
Patients were classified according to high-resolution computed tomography (HRCT) findings and forced vital capacity (FVC) values as pulmonary arterial hypertension (PAH) without ILD, PH-ILD with FVC ⩾70%, or FVC <70%. Survival analysis, clinical outcomes, and prognostic factors were assessed by Kaplan–Meier curves, multivariate logistic regression, and receiver operating characteristic (ROC) analysis.
Results:
Fifty-three patients with SSc-PH were enrolled, and their 5-year overall survival rate was 66.7%. The 29 patients with PH-ILD demonstrated significantly lower survival than the 24 patients with PAH (48% vs 87%, p = 0.005). FVC <70% was associated with markedly reduced survival (29% vs 80% for FVC ⩾70%, p = 0.003). ROC analysis confirmed FVC as a significant prognostic marker (area under the curve = 0.713, p = 0.005). Multivariate analysis identified the FVC <70% at PH diagnosis as the only significant predictor of mortality. Moreover, PH-ILD patients with FVC <70% showed the most significant functional deterioration during follow-up, with a 19% decline in FVC and a greater deterioration in World Health Organization functional class.
Conclusion:
An FVC <70% at baseline represents an important negative prognostic factor in patients with SSc-PH, independently of the extension of ILD on HRCT scan. Functional assessment may complement radiological evaluation and emphasize the importance of routine monitoring of lung function for risk stratification in patients with SSc-PH.
Plain language summary
Systemic Sclerosis is a rare autoimmune disease that can cause high blood pressure in the lungs, leading to serious complications and death. This research study onvestigated 53 scleroderma patients with this lung complication to see if a simple breathing test could predict survival. The breathing test, called forced vital capacity (FVC), measures how much air you can blow out after taking the deepest breath possible. Patients were grouped by their test results: those with FVC above 70% of normal versus those below 70%. After following patients for up to 10 years, the results were striking. Only 29% of patients with poor breathing test results (FVC below 70%) survived 5 years, compared to 80% of those with better results. The breathing test was the single best predictor of survival - even better than expensive CT scans showing lung scarring. This simple, widely available test can help doctors identify high-risk patients who need more intensive monitoring and earlier treatment. For patients with scleroderma, a breathing test result below 70% of normal is a critical warning sign requiring immediate attention and aggressive therapy. The research shows that routine breathing tests should be a key part of care for all scleroderma patients, helping doctors make better treatment decisions and potentially saving lives.
Introduction
Systemic sclerosis (SSc) is a complex autoimmune connective tissue disease characterized by vasculopathy, inflammation, and fibrosis, which affects the skin and various internal organs. 1 Despite its relatively low prevalence, estimated at 50–300 cases per million population, SSc is associated with significant morbidity and mortality. 2
Pulmonary hypertension (PH) is a severe complication of SSc that occurs in approximately 15% of SSc cases. In fact, PH represents one of the leading causes of death in these patients, often leading to a significant deterioration of the quality of life of affected patients, who frequently require supplemental oxygen therapy. 3 SSc patients can develop a spectrum of PH, potentially including all groups defined by the European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines.4,5 Specifically, SSc may be associated with pulmonary arterial hypertension (PAH), categorized as group 1 PH, secondary to pulmonary pre-capillary vascular bed remodeling, leading to increased cardiopulmonary resistance and pressure.4,5 Furthermore, SSc patients may develop PH due to left heart diseases, such as myocardial fibrosis leading to left ventricular dysfunction (Group 2 PH), or lung diseases, such as interstitial lung disease (ILD), which is common in SSc and can contribute to hypoxemia and subsequent PH development (Group 3 PH). Other causes of PH development in patients with SSc include chronic thromboembolic pulmonary hypertension (CTEPH; Group 4 PH) or miscellaneous (Group 5 PH).4,5
Despite significant advances in the understanding of the pathogenetic mechanisms associated with PH development over the past two decades, with the development of specific screening strategies, diagnostic tools, and targeted therapies, the prognosis of SSc-PH remains poor, with reported 3-year mortality rates of approximately 50%.5–7
Furthermore, complicating the management of patients with SSc, the possible subsequent development of ILD may influence the clinical picture of PAH. Using a high-resolution computed tomography (HRCT) scan, ILD, which might not be clinically relevant at the time of PAH diagnosis, may be detected throughout the disease in many patients with SSc. 7 According to recent evidence, the concurrent presence of ILD is the most significant factor affecting PAH treatment response and overall prognosis in patients with SSc.7–9 Interestingly, pulmonary function tests (PFT) may worsen in about 50% of SSc-PAH patients due to ILD development, as confirmed by lung HRCT within the first 3 years from PAH diagnosis, while only a minority of SSc-PAH patients develop a clinically relevant restrictive respiratory syndrome. 9
The functional significance of ILD in patients with SSc affected by PAH and its contribution to overall prognosis have not been clarified. Our study investigated whether forced vital capacity (FVC), as a functional biomarker of ILD burden, may help assess the prognosis of patients with SSc-PH.
Methods
Study population
This study included consecutive SSc patients diagnosed with pre-capillary forms of PH, as Group 1 PH (PAH) or Group 3 PH (PH-ILD), from two Italian outpatient referral centers for SSc between January 2015 and December 2024. Patients were considered eligible if they fulfilled the 2013 European Alliance of Associations for Rheumatology (EULAR)/American College of Rheumatology classification criteria for SSc 10 and had a confirmed diagnosis of pre-capillary PH by right heart catheterization (RHC) according to the available ESC/ERS guidelines for PH since 2015. All enrolled patients underwent RHC echocardiography, chest HRCT, and PFTs at the moment. According to chest HRCT and PFTs results, patients were stratified into three groups:
- Group 1 consisted of patients classified as having PAH without ILD;
- Group 2 comprised patients with PH-ILD, defined as the presence of ILD on HRCT with preserved FVC ⩾70%;
- Group 3 included patients with PH-ILD presenting with severe restrictive respiratory syndrome, characterized by the presence of ILD on HRCT and reduced FVC <70%.5,11
Patients with other forms of SSc-PH, such as those due to left heart disease (Group 2), CTEPH (Group 4), and miscellaneous (Group 5), were excluded from this study.
Data collection
Demographic, clinical, laboratory, and instrumental data were retrospectively collected from medical records at baseline (defined as the time of PAH diagnosis) and during follow-up visits, at least annually, until December 2023. The collected data included age, sex, SSc subtype defined as limited cutaneous or diffuse cutaneous according to the LeRoy classification, 12 disease duration, autoantibody profile (anti-centromere, anti-topoisomerase I), organ involvement, WHO-functional class, and 6-minute walk distance (6 MWD). The FVC and diffusion lung carbon monoxide (DLCO) were collected as the % predicted values at the PFT. The estimated systolic pulmonary artery pressure (sPAP) and tricuspid annular plane systolic excursion were collected from the transthoracic echocardiographic evaluation. All hemodynamic parameters, including mean pulmonary artery pressure (mPAP), pulmonary vascular resistance (PVR), Woods Units, and pulmonary capillary wedge pressure (PCWP), were recorded from the diagnostic right heart catheterization (RHC) reports. Finally, data on pharmacological therapies administered at the time of PH diagnosis and at last follow-up, including corticosteroids (CS), conventional-synthetic disease-modifying antirheumatic drugs (csDMARD), biologic (b)DMARD, nintedanib, calcium channel blockers, diuretics, endothelin receptor antagonists (ERA), phosphodiesterase-5 inhibitors, selexipag, riociguat, and prostanoids, including treprostinil and iloprost, were reported.
The WHO-Functional Class (WHO-FC), typically categorized into four classes from I to IV, 5 was also specified as an intermediate class that cardiologists may have assigned during visits and reported in the patient’s clinical records. Therefore, based on the purpose of our study, WHO-FC has been classified into six classes: 1 (WHO-FC I), 2 (WHO-FC II), 3 (WHO-FC II–III), 4 (WHO-FC III), 5 (WHO-FC III–IV), and 6 (WHO-FC IV). Clinical worsening during follow-up was assumed if a patient experienced worsening WHO-FC, the introduction of oxygen therapy, hospitalization, or death. During follow-up, disease progression was evaluated through periodic clinical examination, WHO-FC assessment, PFT, including changes in FVC and DLCO predicted values, and lung HRCT scan monitoring in all patients with SSc-PH. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement. 13
Risk stratification
The COMPERA 2.0 risk stratification tool, which has been validated in patients with PAH, including those with connective tissue disease, was used to assess mortality risk at baseline and during follow-up, 14 and to guide any therapy adjustments according to the available ESC/ERS guidelines.4,5 The COMPERA 2.0 incorporates the prognostic variables WHO-functional class, 6 MWD, N-terminal pro-brain natriuretic peptide (NT-proBNP) or BNP, right atrial pressure, cardiac index, and mixed venous oxygen saturation to stratify patients at baseline and during follow-up according to their 1-year mortality risk.
Statistical analysis
Descriptive statistics was used to determine the characteristics of the study population. Continuous variables are expressed as mean ± standard deviation (SD) or median and interquartile range, depending on their distribution. Categorical variables are presented as frequencies and percentages. Comparisons between groups were carried out using analysis of variance (ANOVA) with post hoc Bonferroni test for multiple comparisons or U-Mann-Whitney or Chi-square tests when appropriate. Repeated measures ANOVA was performed for time comparisons within groups. Survival rates were estimated using the Kaplan–Meier method, considering death as the outcome of interest and ILD, % predicted FVC value (with 70% as cutoff), or PH categories (PAH and PH-ILD) as exposure variables, using the log-rank test to assess any differences among groups. Multivariate logistic regression analysis was performed to identify predictors of mortality, expressed as odds ratios (OR) with 95% confidence intervals (CI), and included in the model the following variables: sex, age at PAH diagnosis, SSc phenotype subsets, SSc disease duration, interval between SSc and PH diagnosis, presence of Raynaud’s phenomenon, presence of centromeric protein B autoantibody, change in FVC over time, presence of gastrointestinal involvement, risk stratification at diagnosis, and presence of FVC ⩾70%. Receiver operating characteristic (ROC) curve analysis was performed to evaluate the discriminative ability of continuous FVC values as potential prognostic mortality parameters. The area under the curve (AUC) with 95% CI was calculated. Sensitivity and specificity were assessed across different FVC thresholds to determine the optimal cutoff values for prognostic assessment. Missing data were handled using a complete-case approach without imputation. All analyses were performed using the SPSS statistical software, and statistical significance was set at p < 0.05.
Results
Baseline characteristics
Fifty-three patients with SSc and pre-capillary PH were included in the study. The mean age at PH diagnosis was 66 ± 10 years, and most patients were women (88.7%), as shown in Table 1. According to lung HRCT scan findings, 24 (45%) patients had no ILD at the time of RHC and were therefore classified as having PAH, while 29 (55%) had PH-ILD; of these, 15 (51.7%) had baseline FVC ⩾70%, and 14 (48.3%) had restrictive respiratory syndrome (FVC <70%).
Baseline demographics and clinical features of PH-SSc patients stratified by FVC threshold of 70%.

Bold p-values indicate statistical significance (p < 0.05).
PAH versus others.
PH-ILD FVC ⩾70% versus PAH.
PH-ILD FVC <70% versus others.
PH-ILD <70% versus PAH.
PH-ILD <70% versus PAH.
PH-ILD <70% versus PAH.
MWD, 6 min walking distance; CENPB, centromeric protein B; DLCO, diffusion lung carbon monoxide; FVC, forced vital capacity; HRTC, high-resolution computed tomography; ILD, interstitial lung disease; IQR, interquartile range; NSIP, nonspecific interstitial pneumonia; NT-BNP, N-terminal brain natriuretic peptide; PAH, pulmonary arterial hypertension; PAP, pulmonary arterial pressure; PAPm, mean pulmonary artery pressure; PAPs, systolic pulmonary arterial pressure; PCWP, Pulmonary Capillary Wedge Pressure; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; RHC, right heart catheterization; SSc, systemic sclerosis; TAPSE, tricuspid annular plane systolic excursion; UIP, usual interstitial pneumonia; WHO FC, World Health Organization functional class; wo, without.
On lung HRCT scans, the usual interstitial pneumonia pattern was detected in 11% of the patients, while nonspecific interstitial pneumonia was detected in 43% of the cohort, and these were similarly distributed between SSc patients with PH-ILD with or without restrictive respiratory syndrome.
Aside from pulmonary involvement, patients with SSc presented with similar demographic and clinical characteristics. At RHC, no significant differences in mean PAP, PCWP, and PVR were found between PAH patients and group 3 PH patients with or without respiratory restrictive syndrome.
As expected, the mean % predicted FVC was significantly higher in patients with PAH (98.3 ± 15.8) than in those with PH-ILD (75.1 ± 20.3), especially with concomitant restrictive respiratory syndrome (p < 0.001). PH-ILD SSc patients with restrictive respiratory syndrome presented a significantly lower predicted % DLCO (38 ± 12) than those with PAH (50 ± 4; p = 0.05). Moreover, SSc patients with PH-ILD and FVC <70% had a more severe functional impairment with significantly more severe exertional dyspnea defined by WHO-FC (p = 0.005) and 1-year mortality risk based on the assessment with the COMPERA 2.0, stratification tool (p = 0.02).
Regarding therapies, which are extensively described in Table 2, csDMARDs were significantly more frequently prescribed in patients with PH-ILD and FVC ⩾70% (100%) compared to those with PAH (62.5%, p = 0.024), while PAH-targeted therapies, as well as the use of monotherapy, dual therapy, or triple therapy regimens, were similarly distributed among the three groups without significant differences.
Therapy distribution in PH-SSc patients stratified by FVC threshold of 70%.

Data are presented as n (%). p-Values calculated using the Chi-square test comparing the three groups. Bold p-values indicate statistical significance (p < 0.05).
PH-ILD (FVC ⩾70%) versus PAH.
ACEi, angiotensin-converting enzyme inhibitors; ARBs, angiotensin II receptor blockers; bDMARD, biologic disease-modifying antirheumatic drugs; CCB, calcium channel blockers; csDMARD, conventional-synthetic disease-modifying antirheumatic drugs; ERA, endothelin receptor antagonists; FVC, forced vital capacity; PAH, pulmonary arterial hypertension; PDE5i, phosphodiesterase-5 inhibitors; PH-ILD, pulmonary hypertension associated with interstitial lung disease; SSc, systemic sclerosi.
Survival analysis
The 5- and 10-year global survivals were 66.7% and 58.5% in SSc patients with PAH and PH-ILD, respectively, with a median survival of 9.4 (95% CI: 7.4–11.4) years. Patients with PH-ILD had significantly lower survival probabilities than those with PAH (log-rank 8.1, p = 0.005). Specifically, patients with SSc-PAH had a median survival of 9.7 (95% CI: 8.6–10.8) years, PH-ILD patients with FVC ⩾70% had a median survival of 7.6 (95% CI 5.9–9.3) years, and SSc-PH patients with restrictive respiratory syndrome (FVC <70%) had a median survival of 6.8 (95% CI 4.3–9.2; Figure 1).

Survival function with Kaplan–Meier curves in SSc patients with PAH and Group 3 PH with or without restrictive respiratory syndrome (predicted FVC <70%).
Clinical outcomes
During follow-up, clinical worsening was observed in all the patients with PH-SSc. As shown in Figure 2, WHO-FC significantly worsened across the three PH-SSc subgroups over time (PAH, p = 0.01; PH-ILD with FVC ⩾70%, p = 0.01; PH-ILD with FVC <70%, p = 0.003), with the most relevant worsening observed in PH-ILD patients with restrictive respiratory syndrome at study enrollment. Of note, no patient progressed to WHO-FC class III in the PAH group, and in patients with PH-ILD and FVC ⩾70%, no patient overcame WHO-FC class III–IV.

WHO-FC assessment over time in SSc-PH patients with PAH, PH-ILD, and FVC <70% or PH-ILD and FVC ⩾70%.
Pulmonary disease assessment using PFT demonstrated a progressive decline in the predicted % FVC in all PH-SSc subgroups during follow-up, as shown in Figure 3. However, SSc-PH patients with baseline restrictive lung disease presented a significant reduction of the predicted % FVC over time compared to PAH and PH-ILD, with FVC >70% patients, with a steeper decline in the predicted % FVC from a mean (SD) at baseline of 57.6 (17.7)–46.6 (9.1) at the last observation (p = 0.007).
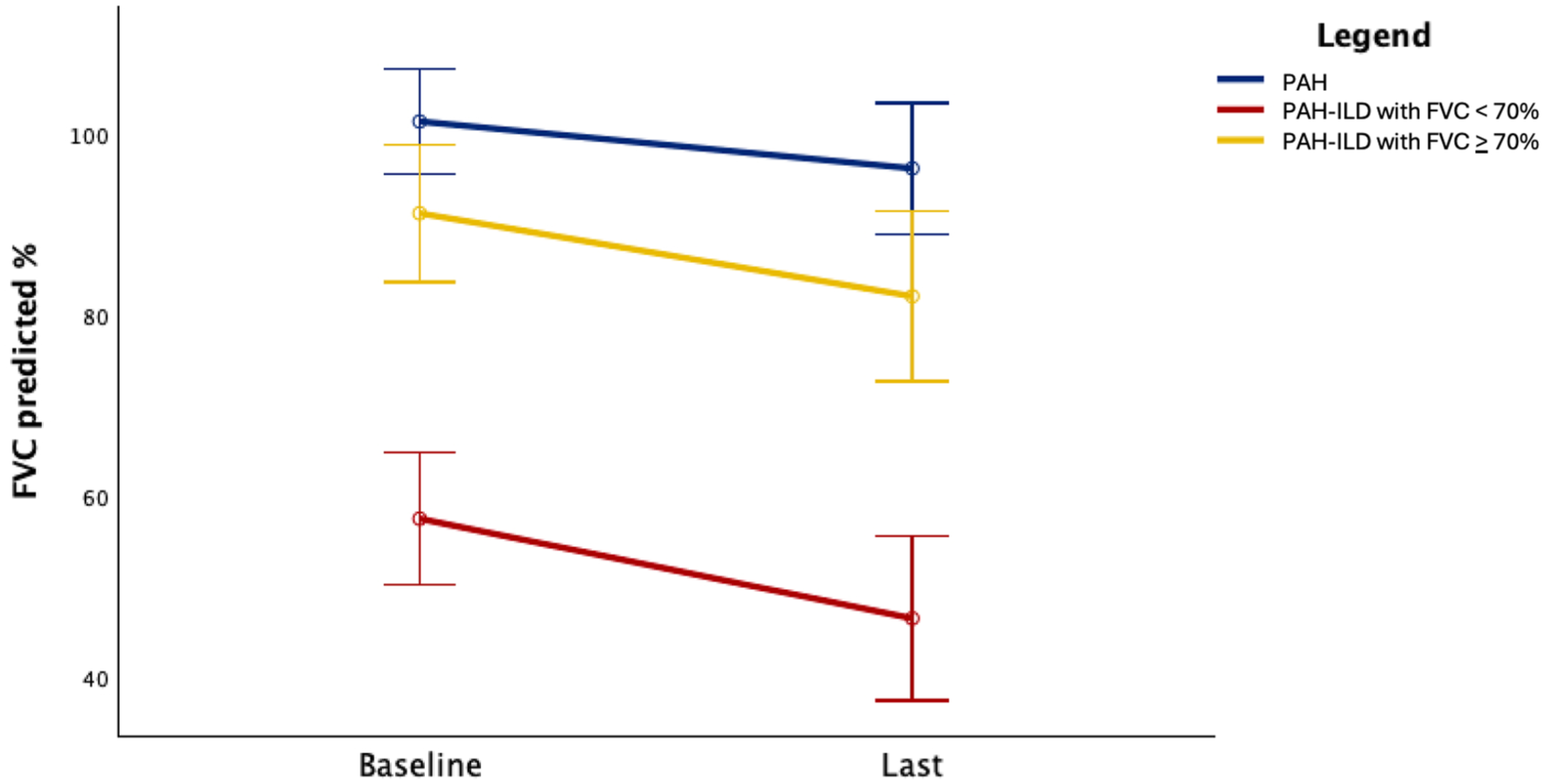
Predicted FVC percent variation over time in SSc-PH patients with PAH, PH-ILD, and FVC <70% or PH-ILD and FVC ⩾70%.
Predictors of mortality
The results of the baseline predictors of mortality estimated by multivariate logistic regression analysis are shown in Table 3. The presence of a predicted baseline FVC ⩾70% was strongly associated with a higher survival probability and was the only negative predictor of mortality (OR = 0.039, 95% CI = 0.002–0.616; p = 0.02). Considering the PH categories as defined in this study (Supplemental Table 1), PH-ILD patients with FVC <70% were the only group presenting a significantly increased risk of death (OR = 68.3, 95% CI: 2.28–145; p = 0.01).
Multivariate logistic regression analysis of mortality predictors in SSc-PH patients, including the variables described below in the model.
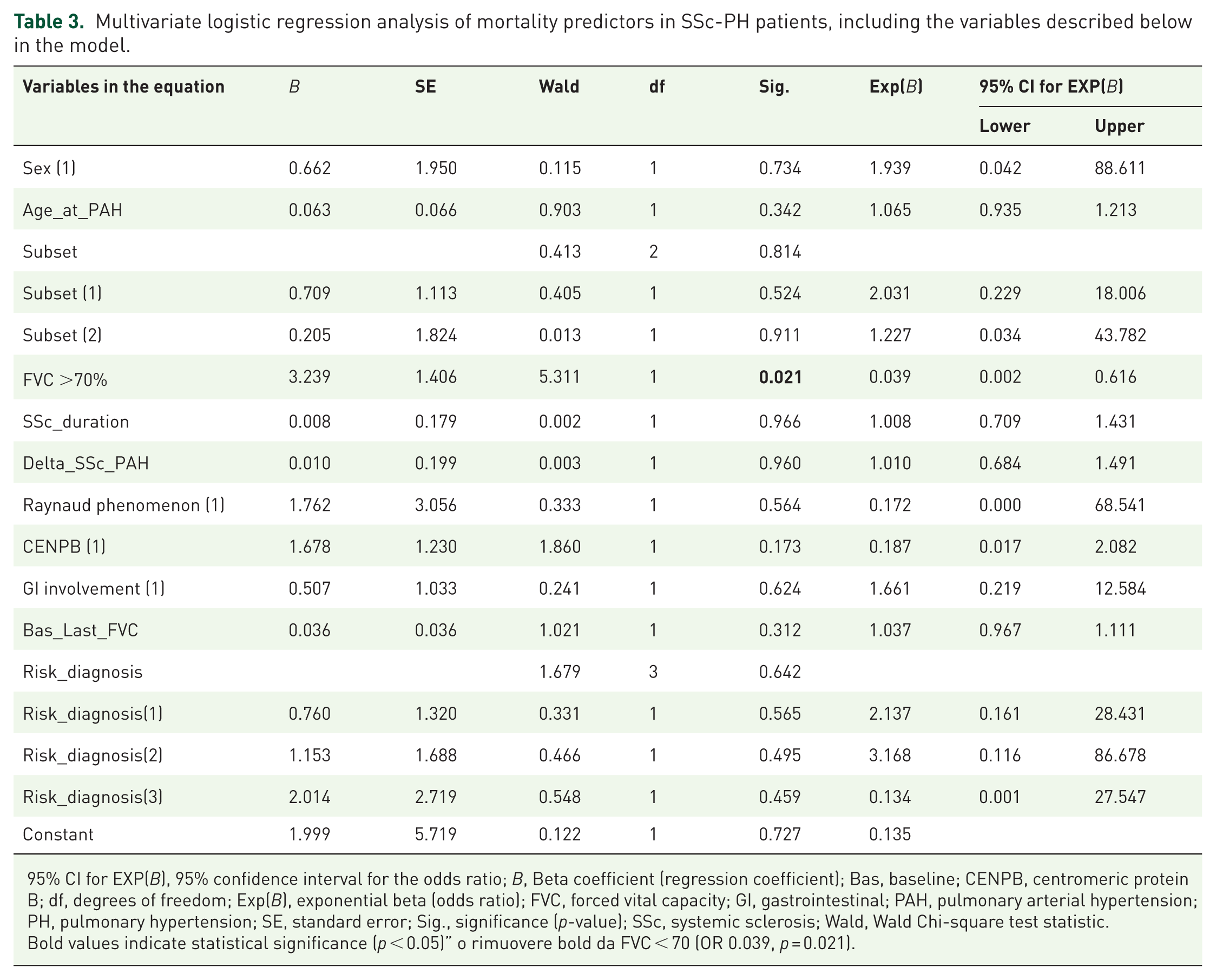
95% CI for EXP(B), 95% confidence interval for the odds ratio; B, Beta coefficient (regression coefficient); Bas, baseline; CENPB, centromeric protein B; df, degrees of freedom; Exp(B), exponential beta (odds ratio); FVC, forced vital capacity; GI, gastrointestinal; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; SE, standard error; Sig., significance (p-value); SSc, systemic sclerosis; Wald, Wald Chi-square test statistic.
Bold values indicate statistical significance (p < 0.05)” o rimuovere bold da FVC < 70 (OR 0.039, p = 0.021).
ROC analysis for FVC as prognostic marker
ROC curve analysis demonstrated that in our cohort, the predicted FVC had moderate discriminative ability as a prognostic parameter (AUC = 0.713, 95% CI: 0.564–0.861; p = 0.005). The predicted % FVC values exhibited a continuous tradeoff between sensitivity and specificity, with an optimal balance of 74.0% as a predictor of mortality (sensitivity, 0.886; specificity, 0.500).
Discussion
This study evaluated the prognostic value of FVC as a functional ILD biomarker in 53 patients from two Italian SSc referral centers. Our key findings demonstrated that the overall 5-year survival rate was 66.7%, with significantly worse outcomes in PH-ILD patients than in PAH patients (48% vs 87%, p = 0.005). Baseline FVC <70% was the strongest independent predictor of poor prognosis (OR = 0.039, p = 0.02), with patients showing markedly reduced survival (29% vs 80% for FVC ⩾70%, p = 0.003). Patients with PH-ILD with FVC <70% demonstrated the most severe functional deterioration during follow-up, including a 19% decline in FVC and greater worsening of WHO-FC. ROC analysis confirmed FVC as a meaningful prognostic marker (AUC = 0.713, p = 0.005), with an optimal threshold of 74%.
Our observed 5- and 10-year survival rates of 66.7% and 58.5%, respectively, were slightly higher than those previously reported in other SSc-PH cohorts. This improvement may reflect advances in screening, early diagnosis, and management of SSc-PH over the past decade.15–17 However, the prognosis of SSc-PAH remains poor compared to that of idiopathic PAH or other connective tissue disease-associated PAH. 18 Moreover, SSc-PAH-associated mortality is even higher in patients with concomitant ILD. 7
According to the most extensive international prospective study on SSc, including 5860 patients from the EULAR Scleroderma Trials and Research cohort, PAH and ILD accounted for most SSc patient deaths. 19 Our findings align with this evidence, demonstrating that the combination of these two complications creates a particularly challenging clinical scenario. In the present study, we investigated whether clinical and instrumental parameters routinely assessed in all SSc patients, including PFT data such as FVC, may predict the prognosis of SSc-PH patients with or without associated ILD. A recent study by Lepri et al. 20 demonstrated a moderate predictive capability of ILD development at the 24-month follow-up in SSc patients with a baseline DLCO <80%, more substantial than a baseline FVC <80%, while the delta changes in the FVC and DLCO did not show a good predictive capability for ILD development. 20 However, it should be considered that while FVC is predominantly a marker of the functioning of the pulmonary parenchyma, DLCO represents a marker of both the pulmonary and vasculature; therefore, changes in the latter may reflect the combined effect of PAH and ILD involvement. 21 Indeed, this study provides compelling evidence that FVC serves as a valuable functional surrogate for the ILD burden in patients with SSc-PH. Our data identified the presence of ILD on lung HRCT and a predicted FVC <70% at the time of PH diagnosis as the main prognostic factors, with evidence of a more pronounced decline during follow-up if the % predicted FVC was already compromised at baseline. Most notably, irrespective of the lung HRCT scan pattern and RHC findings, patients with SSc-PH with a predicted FVC <70% at baseline had the worst survival probability.
These findings are consistent with those of other studies on CTD-associated ILD. In a retrospective monocentric study including 211 Japanese patients with connective tissue disease-associated ILD, an FVC <65% was the only significant mortality predictor in Cox regression analysis, while the specific diagnosis or presence of particular autoantibodies was not a relevant prognostic factor. 22
In contrast, our results contrast with those of a recent Spanish retrospective study by Guillén-Del-Castillo et al., 21 who investigated the impact of ILD on SSc-PAH patient survival. This large registry study of 364 patients with PAH-SSc found that while patients with concomitant ILD presented with worse hemodynamic features and PFTs, there was no significant difference in transplant-free survival compared to those without ILD. Notably, up-front combination therapy was used in approximately 60% of the patients and was independently associated with improved survival. The discrepancy between their findings and ours may reflect differences in patient populations, treatment approaches, or the potential for aggressive PAH-targeted treatment to overcome the negative prognostic impact of ILD.
Clinical impairment defined by the WHO-FC showed a faster progression of symptoms in patients with PH-ILD with concomitant restrictive syndrome at baseline. Interestingly, during follow-up, the deterioration of % predicted FVC in SSc patients with baseline FVC <70% was twice that of patients with baseline FVC ⩾70%, demonstrating a more rapid progression of damage in SSc-PH patients with concomitant restrictive respiratory syndrome. These findings confirm the clinical challenge of treating patients with SSc-PH with a progressive ILD phenotype, requiring more careful assessment and management, with timely use of currently available antifibrotic agents. 23 The accelerated functional decline in patients with compromised FVC suggests that early intervention may be crucial in this high-risk subgroup. Our ROC analysis results supported the use of FVC as a meaningful prognostic marker (AUC = 0.713, 95% CI: 0.564–0.861, p = 0.005). This revealed that FVC, used as a continuous parameter, presented optimal sensitivity and good specificity at a threshold of 74%, slightly higher than the 70% commonly considered discriminatory value for restrictive pulmonary disease. 24 Identification of FVC <70% as the primary adverse prognostic factor has important clinical implications. This simple and widely available parameter can aid in risk stratification at the time of PH diagnosis and guide treatment decisions. Patients with FVC <70% may benefit from more aggressive monitoring, earlier initiation of combination therapies, and consideration of anti-fibrotic treatment.
Limitations of the study
The sample size of this study was limited and not pre-calculated through a power analysis. Specifically, considering that SSc is a rare disease and that less than 15% of patients develop PH as a complication, all SSc-PH patients with available data and meeting the inclusion criteria of this study were included in the analysis. Another limitation of this study is its retrospective design, with the occurrence of missing data potentially dampening the strength of our results.
Despite these limitations, our results highlight the importance of routine pulmonary function monitoring in all patients with SSc. This plays a crucial role in SSc patients with concomitant Group-3 PH at diagnosis, for whom a higher risk of progressive ILD deterioration could be estimated, with the need for closer surveillance and timely initiation of appropriate antifibrotic therapy.25,26
Conclusion
Our study highlights the importance of adequate screening and early diagnosis of PH with potential pulmonary involvement using PFT and lung HRCT with close monitoring and timely specific treatment adjustments to impact the survival probabilities of SSc-PH patients. A low baseline % predicted FVC (<70%) was identified as the primary adverse prognostic factor in SSc patients with Group 3 PH. Regardless of the extent of impairment on lung HRCT scan, the functional loss evidenced by % predicted FVC at the diagnosis of PH significantly compromises the prognosis of SSc patients.
Further research is needed to validate our results in larger cohorts and to investigate whether the identified use of the pathological FVC cutoff can aid risk stratification and diagnostic–therapeutic decision-making.
Supplemental Material
sj-docx-1-tab-10.1177_1759720X251410681 – Supplemental material for Forced vital capacity as a survival predictor in systemic sclerosis-associated pulmonary hypertension
Supplemental material, sj-docx-1-tab-10.1177_1759720X251410681 for Forced vital capacity as a survival predictor in systemic sclerosis-associated pulmonary hypertension by Claudia Iannone, Stefano Stano, Maria Rosa Pellico, Andrea Cito, Marco Capodiferro, Roberto Caporali, Fabio Cacciapaglia and Nicoletta Del Papa in Therapeutic Advances in Musculoskeletal Disease
Supplemental Material
sj-pdf-2-tab-10.1177_1759720X251410681 – Supplemental material for Forced vital capacity as a survival predictor in systemic sclerosis-associated pulmonary hypertension
Supplemental material, sj-pdf-2-tab-10.1177_1759720X251410681 for Forced vital capacity as a survival predictor in systemic sclerosis-associated pulmonary hypertension by Claudia Iannone, Stefano Stano, Maria Rosa Pellico, Andrea Cito, Marco Capodiferro, Roberto Caporali, Fabio Cacciapaglia and Nicoletta Del Papa in Therapeutic Advances in Musculoskeletal Disease
Footnotes
Acknowledgements
The authors thank all patients who participated in this study and the clinical staff of both participating centers.
Declarations
Supplemental material
Supplemental material for this article is available online.
Artificial intelligence policy
The authors declare that they have not used any generative artificial intelligence (AI; e.g., ChatGPT) in creating text, references, images, or other content.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.