
Abstract
Background:
In patients with rheumatoid arthritis (RA), persistent inflammation and increasing disease activity are associated with increased risk of adverse events (AEs).
Objectives:
To assess relationships between RA disease activity and AEs of interest in patients treated with tofacitinib or tumor necrosis factor inhibitors (TNFi).
Design:
This was a post hoc analysis of a long-term, postauthorization safety endpoint trial of tofacitinib versus TNFi.
Methods:
In ORAL Surveillance, 4362 patients aged ⩾50 years with active RA despite methotrexate, and ⩾1 additional cardiovascular (CV) risk factor, were randomized 1:1:1 to tofacitinib 5 or 10 mg twice daily or TNFi for up to 72 months. Post hoc time-dependent multivariable Cox analysis evaluated the relationships between disease activity [Clinical Disease Activity Index (CDAI)], inflammation [C-reactive protein (CRP)], and AEs of interest. The AEs included major adverse CV events (MACE), malignancies excluding nonmelanoma skin cancer (NMSC), venous thromboembolism (VTE), serious infections, herpes zoster (HZ), nonserious infections excluding HZ (NSI), and death.
Results:
Across treatments, risk for NSI was higher when patients had CDAI-defined active disease versus remission; MACE and VTE risks trended higher, but did not reach significance. Hazard ratios for MACE, malignancies excluding NMSC, VTE, infections, and death rose by 2–9% for each 5-mg/L increment in serum CRP. The interaction terms evaluating the impact of treatment assignment on the relationship between disease activity and AEs were all p > 0.05.
Conclusion:
In ORAL Surveillance, higher NSI risk was observed in the presence of active RA versus remission. The risk of MACE and VTE directionally increased in active disease versus remission, although statistical power was limited due to small event numbers in these categories. The relationship between active disease and AEs was not impacted by treatment with tofacitinib versus TNFi.
Registration:
NCT02092467.
Plain language summary
• People with rheumatoid arthritis (RA) who have uncontrolled symptoms (high disease activity) have a higher chance of having adverse medical events (medical problems that occur during treatment with a medication) than people who have mild symptoms (low disease activity).
• We looked at the link between levels of disease activity and the risk of having adverse medical events in people with RA who took tofacitinib or a tumor necrosis factor inhibitor (TNFi) medication.
• We used the results of ORAL Surveillance, a long-term safety trial in people with RA.
○ In this study, people with RA were 50 years or older and at high risk of a major cardiovascular event such as heart attack or stroke.
• For up to 6 years, people took tofacitinib 5 or 10mg tablets two times a day or TNFi injections.
• We used statistical tests to examine the link between different levels of RA disease activity or inflammation and different adverse medical events, such as:
○ major cardiovascular events (such as heart attack, stroke, or death due to heart failure)
○ cancers
○ blood clots
○ infections
○ deaths.
• In people who took tofacitinib or TNFi:
○ People with active disease (those with RA symptoms) had a higher risk of infections that did not lead to hospitalization (nonserious infections) than people in remission (those with very mild symptoms or no symptoms at all).
○ People with active disease also had a slightly higher risk of major cardiovascular events and blood clots than those in remission.
○ Higher levels of inflammation led to increased risk of major cardiovascular events, cancers, blood clots, infections, and deaths.
• Active RA disease leads to higher risk of adverse medical events.
• The medication used (tofacitinib or TNFi) did not affect the link between levels of RA disease activity and adverse medical events.
• This study was limited by the low number of adverse medical events recorded.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by synovial inflammation and the potential for joint destruction. 1 While RA disease activity is primarily measured based on the extent of joint involvement and patient-reported outcomes, RA-related inflammation is systemic. Continued inflammation and increasing disease activity are associated with increased risk of comorbidities and adverse events (AEs),1,2 including major adverse cardiovascular (CV) events (MACE),3–10 venous thromboembolism (VTE), 11 malignancies,12–15 and both serious and nonserious infections (NSIs).16–18
Recognizing the potential hazards associated with uncontrolled inflammation on both joints and extra-articular tissues, the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) have incorporated treat-to-target recommendations into their treatment guidelines.19,20 These state that either low disease activity (LDA) 19 or clinical remission should be the primary target for the treatment of RA20,21; LDA may be more realistic among patients with long-standing disease. 21 The availability of multiple advanced therapies with different mechanisms of action increasingly enables attainment of these therapeutic goals. 22 This may in turn mitigate the risk of AEs caused by higher disease activity. For instance, in an integrated post hoc safety analysis of the SELECT phase III RA clinical program of upadacitinib, patients who received upadacitinib 15 mg and experienced MACE or VTE showed lower improvement in their disease activity versus patients who did not have an event. 23
ORAL Surveillance (NCT02092467) was a large, long-term, clinical safety endpoint-driven, postauthorization trial of tofacitinib versus tumor necrosis factor inhibitors (TNFi) in a CV risk-enriched patient population with coprimary endpoints of MACE and malignancies excluding nonmelanoma skin cancer (NMSC). 24 The size and duration of this study enabled the evaluation of a broad range of AEs, including those that occur at low incidence and/or long latency, within a single cohort of patients in a prospective, randomized setting. In this CV risk-enriched population, risks of MACE and malignancies were higher with tofacitinib versus TNFi and did not meet noninferiority criteria. 24
In this post hoc analysis of ORAL Surveillance, we sought to characterize the relationship between RA disease activity and AEs of interest, after adjusting for other baseline risk factors. We also evaluated whether tofacitinib or TNFi treatment had any impact on the relationship between disease activity and AE risk.
Methods
Study design and patients
ORAL Surveillance was a randomized, open-label, noninferiority, postauthorization phase 3b/4 safety endpoint-driven study that enrolled patients with active RA despite methotrexate treatment aged ⩾50 years with ⩾1 additional CV risk factor. All patients satisfied the 2010 ACR/EULAR Classification Criteria for RA. 25
Patients were randomized 1:1:1 to receive oral tofacitinib 5 or 10 mg twice daily (BID), or subcutaneous TNFi [adalimumab 40 mg once every 2 weeks (North America: US, Puerto Rico, and Canada) or etanercept 50 mg once weekly (rest of the world)]. All patients continued to receive background methotrexate, unless modification was clinically indicated. 24
The study began enrollment in March 2014 and completed in July 2020; the database was released in December 2020. In February 2019, the tofacitinib 10 mg BID dose was reduced to 5 mg BID after the Data Safety Monitoring Board noted an increased frequency of pulmonary embolism (PE) in patients receiving tofacitinib 10 mg BID versus TNFi, and an increase in overall mortality with tofacitinib 10 mg BID versus 5 mg BID and TNFi. Data were collected for up to 72 months of follow-up. Patients randomized to the tofacitinib 10 mg BID group whose dose was reduced to 5 mg BID, or who discontinued from the trial drug, were included in the tofacitinib 10 mg BID group.
Endpoints
This post hoc analysis evaluated the relationship between RA-related inflammation (time-dependent, measured at baseline and postbaseline) and selected AEs of interest. Clinical Disease Activity Index (CDAI) and C-reactive protein (CRP) were used as measures of RA-related disease activity and inflammation. CDAI incorporates assessments of tender and swollen joint counts (28 joints assessed), and both patient’s and physician’s global assessments of disease activity; CRP is routinely assessed as a marker of systemic inflammation in RA. 26 A total of 14 AEs were included: adjudicated MACE (along with its components of myocardial infarction and stroke); adjudicated malignancies (three endpoints: malignancies excluding NMSC, NMSC, and lung cancer); adjudicated VTE [three endpoints: all VTE, deep vein thrombosis (DVT), and PE]; serious infection (defined as any event in the Infections and Infestations System Organ Class, marked as serious); herpes zoster (HZ; nonserious and serious); NSI excluding HZ; and adjudicated death (two endpoints: all-cause deaths and deaths due to CV events).
Statistical analysis
Safety endpoints were analyzed using the safety analysis set, which included all randomized patients who received ⩾1 dose of study treatment. Patients, including those required to switch from tofacitinib 10 mg BID to 5 mg BID in February 2019, were analyzed in their originally randomized group; the data collected after the dose switch were counted in the tofacitinib 10 mg BID group, as described previously. 24
Safety events were counted within predefined risk periods and censoring times, per the ORAL Surveillance study protocol and statistical analysis plan. 24 For MACE and its components, the risk period was defined as the time from the first dose until the last contact date or the last trial dose plus 60 days, whichever was earliest. For malignancies excluding NMSC and lung cancer, the risk period was the time from the first dose until the last contact date. For all other endpoints (NMSC, VTE, infections, and death), the risk period was the time from the first dose until the last contact date or the last trial dose plus 28 days, whichever was earliest.
For each endpoint indicated above, our model-building approach to assessing relationships between RA-related inflammation and the safety events consisted of the following multiple steps. First, potential baseline risk factors were selected based on clinical knowledge and literature.6,27–29 These included patient demographics, medical history, RA disease characteristics, and baseline and prior medications (the full list of covariates are listed in the Supplemental methods). Second, we performed simple Cox analyses for each baseline risk factor (including treatment arm and a single candidate risk factor), one at a time. Any baseline risk factor with a p < 0.10 from the simple Cox models was selected by the model. Third, a multivariable Cox proportional hazard model analysis using backward selection was performed, including treatment arm (always included) and all the variables that had been selected by the model in the second step. The p value cutoff for a risk factor to stay in the multivariable model was 0.10 (i.e. p < 0.10). Finally, time-dependent CDAI categories were added to this multivariable model for each AE of interest that had been selected in the third step. CDAI categories were defined as low (>2.8 and ⩽10; LDA), moderate (>10 and ⩽22; MDA), or high (>22; HDA) disease activity versus remission (⩽2.8). Time-dependent CDAI categories included all baseline and postbaseline CDAI values recorded for each patient prior to the AE of interest for patients who experienced the AE, or the end of the risk period for patients who did not experience the AE. The counting process method of data input was used, that is, the time was first divided into intervals at cut points where CDAI categories were evaluated; then, the presence or absence of the AE was counted in each interval (event interval or censored interval, respectively) for which the beginning CDAI category was the covariate for that interval. 30 This models the entire disease trajectory based on CDAI of the patient during the study. Hazard ratios for LDA, MDA, and HDA versus remission and 95% confidence intervals (CIs) were estimated based on this multivariable time-dependent Cox proportional hazard model. 31 Nominal p values <0.05 were considered evidence of associations between categorized CDAI and risk of AEs. This analysis was similarly repeated for time-dependent CRP (mg/L) as a continuous covariate, with hazard ratios estimated per increment of 5 mg/L of CRP. Pooling of the AEs across the two tofacitinib and TNFi arms enhanced the precision of the estimates, particularly for less frequently occurring AEs.
Separately, a sensitivity analysis including the interaction between treatment arm and the time-dependent covariate was performed to assess whether the effect of CDAI (or CRP) on the risk of AE was different for tofacitinib versus TNFi.
In addition, cumulative disease activity (per year) was calculated using the area under the curve (AUC) for CDAI from baseline to the time of the AE. Comparisons of the CDAI AUC/year for patients with versus without AEs were made using an analysis of variance model with treatment arm, event status, and the interaction between treatment arm and event status as covariates.
Across these analyses, no multiplicity adjustments were applied.
Results
Patients
Overall, 4362 patients were included (tofacitinib 5 mg BID: N = 1455; tofacitinib 10 mg BID: N = 1456; TNFi: N = 1451) with a median follow-up of 4.0 years. Detailed demographics and baseline characteristics have been previously reported 24 ; selected characteristics are shown in Table 1. Across treatment arms, 78.2% of patients were female; median age was 60 years. Mean disease duration at baseline was 10.4 years. By study design, patients were receiving concomitant methotrexate at baseline; 28.4% had previously received another conventional synthetic disease-modifying antirheumatic drug (DMARD) (csDMARD) and 10.4% a biologic DMARD (bDMARD). Furthermore, 57.2% of patients reported corticosteroid use at baseline. Baseline CDAI scores and CRP levels were similar across treatment arms (Table 1).
Baseline patient demographics and characteristics.
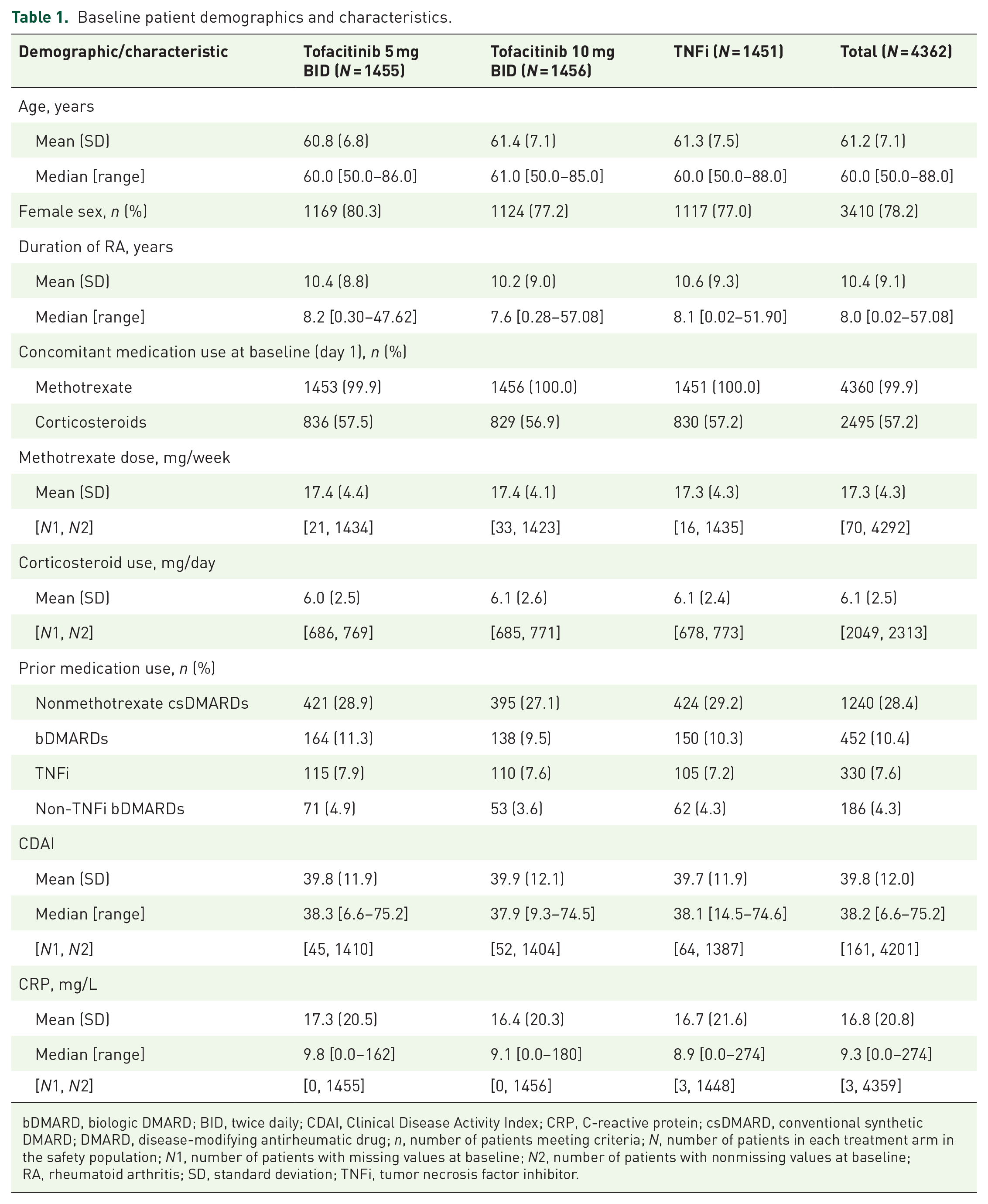
bDMARD, biologic DMARD; BID, twice daily; CDAI, Clinical Disease Activity Index; CRP, C-reactive protein; csDMARD, conventional synthetic DMARD; DMARD, disease-modifying antirheumatic drug; n, number of patients meeting criteria; N, number of patients in each treatment arm in the safety population; N1, number of patients with missing values at baseline; N2, number of patients with nonmissing values at baseline; RA, rheumatoid arthritis; SD, standard deviation; TNFi, tumor necrosis factor inhibitor.
Patients were assessed for disease activity, as defined by CDAI, at every study visit. Improvements in CDAI were observed in all three treatment arms. Figure 1 graphically depicts the proportions of patients in each disease activity category by visit, for the duration of the study across all treatment arms. CDAI category changes during the study were similar across treatment arms, as shown in Supplemental Figure S1.

Patients in CDAI-defined disease activity categories in ORAL Surveillance, by visit (all treatments).
The results of the Cox regression analysis showed that, at any given point in time during the study, patients with residual disease activity (CDAI HDA, MDA, or LDA) had an increased risk of certain AEs versus patients in remission. This residual disease activity was a risk factor for NSI. For MACE, VTE, serious infection (MDA and LDA only), and CV death (MDA only), patients with active disease trended toward a higher risk of developing these AEs versus those in remission, but all p values were >0.05 (Figure 2). No association with CDAI category was observed for malignancies or HZ (Figure 2; Supplemental Figure S2). Results of additional endpoints, including components of MACE, malignancy subtypes, DVT, and PE, are shown in Supplemental Figure S2.

Hazard ratios (95% CIs) for selected AEs of interest: CDAI-defined active RA versus remission (time-dependent covariate analysis).
Additionally, a sensitivity analysis indicated that the relationship between CDAI and AEs was not impacted differentially by treatment arm (all interaction terms had p > 0.05; data not shown).
Higher serum CRP was associated with greater risk for most AEs of interest
Results from the Cox regression analysis showed that risks of AEs of interest increased with higher serum CRP, except for HZ (Figure 3). Specifically, risk increased, with each 5 mg/L increment in serum CRP, by 6% (95% CI: 2–9%) for MACE, 4% (2–6%) for malignancies excluding NMSC, 5% (0–10%) for VTE, 9% (7–10%) for serious infections, 2% (1–3%) for NSI, and 9% (4–13%) for CV death (all p < 0.05; Figure 3). In contrast, there was a 6% (0–11%) reduction in risk for HZ for each 5-mg/L increment in serum CRP. Results of analyses for additional endpoints, including components of MACE, VTE, and malignancies, are shown in Supplemental Figure S3. The sensitivity analysis again showed that the relationship between CRP and AEs was not impacted differentially by treatment arm (data not shown; all interaction p values > 0.05).

Hazard ratios (95% CIs) for selected AEs of interest: 5 mg/L CRP increment (time-dependent covariate analysis).
Patients who experienced certain AEs had greater cumulative CDAI disease activity
Comparison of least squares mean AUC/year for CDAI showed that patients who received tofacitinib 5 mg BID and experienced MACE, VTE, serious infections, and NSI had higher cumulative disease activity exposure during the study than patients on the same treatment who did not experience these events (all p values <0.05; Supplemental Table S1). Patients who received tofacitinib 10 mg BID and experienced VTE, serious infections, HZ, and NSI also experienced higher cumulative disease activity than patients on the same treatment who did not experience these events (all p values <0.05; Supplemental Table S1). For patients who received TNFi, those who experienced serious infections and NSI had higher cumulative disease activity per year than patients who did not experience infection events (all p values <0.05; Supplemental Table S1).
Discussion
RA is associated with a range of comorbid conditions such as infections and CV disease.1,2 In addition to providing valuable information about the risk profile of tofacitinib versus TNFi, ORAL Surveillance also allowed an opportunity to explore risk factors for many AEs of interest in patients with RA who were receiving active treatment. At the time of enrollment, participants in this trial were substantially undertreated by treat-to-target standards and practice guidelines. 20 This was evidenced by the findings that patients in this trial had mean disease duration of 10 years at enrollment and, although they were receiving methotrexate at baseline (by protocol), less than 30% had received other csDMARDs previously, indicating a low rate of combination csDMARD therapy, and approximately 10% had received a prior bDMARD. However, over half of these patients were receiving corticosteroids at the time of enrollment. In spite of this, approximately 96% of patients entered the study with a CDAI score consistent with HDA. We propose that not only were these patients enriched for AEs of interest due to age and comorbid CV risk factors, but also due to their HDA at enrollment, and their historically undertreated RA.4,8,13,15,16,18 Although ORAL Surveillance was a long study, with a mean patient follow-up of 4 years, one limitation of this analysis was that we were not able to assess the cumulative burden of inflammation with which the patient entered the study because the clinical trial did not allow for retrospective capture of the CDAI and CRP values for the patients’ entire duration of disease, which ranged from <1 month to 57 years.
Focusing on the disease activity measures during the study, we used a Cox regression model with time-dependent CDAI or CRP to explore relationships between patients’ CDAI score or CRP level and the risk of incurring AEs. The analysis included patient demographics, medical history, RA disease characteristics, and baseline medications. In this high-risk population of patients with RA, residual disease activity was a contributory risk for AEs. For instance, NSI risks were higher among patients with active RA than those in remission, and risk increased with rising CDAI disease activity. The analysis also suggested that MACE and VTE risks trended higher when patients had active disease versus remission, but the p values were >0.05, and a similar increase with higher CDAI activity was not demonstrated. Separately, we found that higher CRP values were associated with a greater risk for MACE, malignancies, VTE, serious infections, NSI, and CV death. It should be noted that at least ~30% of patients had MDA/HDA throughout the duration of study, suggesting that the relationship between disease activity and AE risk may have been observed due to patients not attaining the treatment target; this is despite the likelihood of patients exhibiting improvements from baseline in CDAI with tofacitinib or TNFi treatment. 32
In the sensitivity analyses, the lack of interactions between treatment arm and CDAI or CRP suggests that disease activity is a risk factor for the AEs of interest, independent of treatment with tofacitinib or TNFi. However, the sensitivity analysis was limited by low power, owing to a generally low number of AEs in the tofacitinib and TNFi arms. Nevertheless, postbaseline CRP values may have been differentially impacted by treatment. Moreover, this analysis assumed that CRP values were related to RA disease activity, although CRP is a nonspecific measure of inflammation and can also fluctuate due to RA-independent factors such as infection and injury. 33 Finally, the CDAI and CRP values included in this analysis were those collected at prescheduled study visits every 3 months from baseline to the visit before the event, rather than when the patient was seen for the AE (e.g. in the emergency room). It is possible that CRP levels fluctuated due to undiagnosed but developing AEs, and the direction of the relationship between CRP and AEs should thus be interpreted with caution.
One seemingly paradoxical finding in our analysis was that, while higher time-dependent CRP was associated with both serious infections and NSI, the relationship was reversed for HZ. The finding that patients receiving Janus kinase inhibitors, including tofacitinib, are at a greater risk for HZ infection has been seen across multiple datasets.34–36 It is plausible that patients who responded best to treatment were also at the highest risk for HZ. However, identifying a mechanistic explanation for this finding requires further studies.
In the AUC/year analysis, we observed that patients receiving tofacitinib 5 mg BID who experienced MACE, VTE, serious infections, or NSI were exposed to higher cumulative disease activity (CDAI) than patients who received the same treatment and did not experience such an event. Similar results were obtained in patients who received TNFi and experienced serious infections and NSI versus patients who did not experience such an event. However, a similar relationship between disease activity and malignancy (excluding NMSC) or CV death was not detected. As with the sensitivity analyses, these analyses were limited by low power, due to the low number of AEs across treatment arms.
Overall, our findings regarding the contribution of inflammation to the risk of AEs in patients with RA are consistent with those of others. Baseline CRP has been highlighted as a predictor of CV death in patients with new onset inflammatory polyarthritis who were followed for approximately 10 years 37 ; similarly, CRP is also an independent predictor of coronary atherosclerotic plaque progression in patients with RA. 4 Two prior studies showed that the risk of MACE increases with acute flares of RA disease.7,8 Importantly, a recent study suggested that the relationship between CDAI and CV risk was the strongest in the first 6 months of therapy and that the association diminished over time 38 ; it is, therefore, possible that the inclusion of all CDAI values over the period of this 5-year study may have attenuated the relationship between disease activity and MACE.
ORAL Surveillance was powered for comparisons between two active therapies, tofacitinib and TNFi, for MACE and malignancies excluding NMSC as primary endpoints; the trial was not powered to assess other endpoints, and it did not include a placebo arm. Our analysis showed a statistical association between active disease by CDAI and NSI, the most frequently occurring AE of special interest in this study. This was the most statistically robust analysis in terms of power. This is consistent with previous findings; a large US cohort study described an association between increasing disease activity and outpatient infection rates among patients with RA. 16 The remaining endpoints in our analysis had low event numbers, and resultant large CIs, potentially explaining some aberrant findings across residual disease activity categories versus remission. Nevertheless, even for AEs with small event numbers, like VTE, the relationship between disease activity and events was still directionally aligned with previous studies. For example, it has been shown that inflammation in RA can contribute to increased risk for VTE, including a long-term registry-based study noting a strong association between increasing acute RA disease activity, measured by Disease Activity Score in 28 joints erythrocyte sedimentation rate, and the risk of VTE. 11 The majority of PE cases reported in ORAL Surveillance were identified in isolation, without knowledge of when the primary thrombosis (presumed DVT) had occurred, potentially impacting the identification of time-dependent disease activity as it is related to the temporal diagnosis of PE.
In this study, we did not find associations between CDAI and malignancies, but CRP was associated with malignancies, which as previously stated may have been due to the malignancy itself rather than RA activity. Lymphoma has the most frequently reported link between lifetime exposure to inflammation and malignancies in patients with RA in existing literature.14,15 Our analysis did not include lymphoma as a standalone endpoint due to insufficient case numbers (four patients receiving tofacitinib 5 mg BID, six patients receiving tofacitinib 10 mg BID, and one patient receiving TNFi). 39 Lymphoma was included in the malignancies excluding NMSC outcome.
Our study was limited by its post hoc nature. ORAL Surveillance included a CV risk-enriched population of patients with RA (aged ⩾ 50 years, with ⩾1 additional CV risk factor); extrapolating our results to the general RA patient population should be done with caution. Associations identified by Cox analyses do not necessarily imply causation, and time-dependent covariate analyses may introduce bias in terms of reverse causality. Backward selection, while commonly used in analyzing clinical trial data, 40 may yield a biased relationship between selected covariates and the outcome; CIs and p values may be underestimated. 41 In addition, the power for the interaction analyses (effect modification) was low. Further, the stability of the backward selection may be affected by a small number of events in some instances. 40 Patient demographics were captured only at baseline; given the long study duration, demographics and thus risk factors (e.g. weight, smoking status, and concomitant medications) may have changed during the study. Due to the conditional nature of the selection method for model building, inference regarding association from the models thus selected is not confirmatory and would warrant further investigation.
Treat-to-target recommendations for the management of RA establish remission as the ideal goal, while LDA may be an acceptable alternative in certain patients.19,20 Our results indicate that in patients with RA ⩾50 years of age with ⩾1 additional CV risk factor, any residual disease activity can contribute to increased risk of certain AEs, despite treatment with advanced immunomodulatory therapies. The risk imposed by disease activity appears to be similar for patients receiving tofacitinib and TNFi. This may suggest that in patients with comorbidities, such as the ORAL Surveillance population, exposure to even modest levels of inflammation may increase the risk of AEs, in line with recent literature.4,11 Clinicians should consider stringent adherence to treat-to-target guidelines in this high-risk patient population to achieve and maintain tight control of disease activity.
Supplemental Material
sj-docx-1-tab-10.1177_1759720X231201047 – Supplemental material for Rheumatoid arthritis disease activity and adverse events in patients receiving tofacitinib or tumor necrosis factor inhibitors: a post hoc analysis of ORAL Surveillance
Supplemental material, sj-docx-1-tab-10.1177_1759720X231201047 for Rheumatoid arthritis disease activity and adverse events in patients receiving tofacitinib or tumor necrosis factor inhibitors: a post hoc analysis of ORAL Surveillance by George A. Karpouzas, Zoltán Szekanecz, Eva Baecklund, Ted R. Mikuls, Deepak L. Bhatt, Cunshan Wang, Gosford A. Sawyerr, Yan Chen, Sujatha Menon, Carol A. Connell, Steven R. Ytterberg and Mahta Mortezavi in Therapeutic Advances in Musculoskeletal Disease
Footnotes
Acknowledgements
The authors would like to thank All-shine Chen, Harry Shi, Jose Rivas, and Joseph Wu from Pfizer Inc for their guidance in the development of this manuscript. Medical writing support, under the direction of the authors, was provided by Karen Thompson, PhD, CMC Connect, a division of IPG Health Medical Communications and was funded by Pfizer, New York, NY, USA, in accordance with Good Publication Practice (GPP) guidelines (Ann Intern Med 2022; 175: 1298–1304). Editorial assistance with submission was provided by Eva Dekker, CMC Connect, IPG Health Medical Communications, and was funded by Pfizer, New York, NY, USA. The authors authorized the submission of their manuscript via a third party and approved all statements and declarations, for example, conflicts of interest and funding.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.