
Abstract
Autoinflammatory diseases are disorders of the innate immune system, which can be either monogenic due to a specific genetic mutation or complex multigenic due to the involvement of multiple genes. The aim of this review is to explore and summarize the recent advances in pathogenesis, diagnosis, and management of genetically complex autoinflammatory diseases, such as Schnitzler’s syndrome; adult-onset Still’s disease; synovitis, acne, pustulosis, hyperostosis, osteitis syndrome/chronic recurrent multifocal osteomyelitis/chronic non-bacterial osteomyelitis; Adamantiades-Behçet’s disease; Yao syndrome; and periodic fever with aphthous stomatitis, pharyngitis, and adenitis syndrome. The PubMed database was screened for relevant articles using free text words and specific search strings. The search was limited to English-language articles, reporting the results of studies in humans, published through March 2021. Evidence from literature suggest that these rare multigenic autoinflammatory diseases can present with different clinical features and the diagnosis of these diseases can be challenging due to a combination of nonspecific manifestations that can be seen in a variety of other conditions. Diagnostic delays and disease complications may occur due to low disease awareness and the lack of pathognomonic markers. The pathogeneses of these diseases are complex and in some cases precise pathogenesis is not clearly understood. Conventional treatments are commonly used for the management of these conditions, but biologics have shown promising results. Biologics targeting proinflammatory cytokines including IL-1, IL-6, TNF-α, IL-17A and IL-18 have been shown to ameliorate signs and symptoms of different multigenic autoinflammatory diseases.
Keywords
Introduction
Autoinflammatory diseases are disorders of the innate immune system characterized by severe stimulation of inflammatory pathways without involvement of antigen-directed autoimmunity. Over the last 20 years, there have been notable developments in the field of autoinflammation and research in this area has led to increased understanding of disease mechanisms and management of these conditions. 1 Autoinflammatory diseases can be either monogenic or multigenic (polygenic). In monogenic autoinflammatory syndromes, a specific genetic mutation can be identified, primarily due to errors in the innate immune mechanism. 2 Multigenic autoinflammatory syndromes are not associated with a single, identified genetic mutation and their genetic characterization is complex due to the involvement of multiple genes. Certain multigenic autoinflammatory syndromes may present more frequently in adults such as Schnitzler’s syndrome and Yao syndrome while other syndromes such as periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome and chronic recurrent multifocal osteomyelitis (CRMO) may occur frequently in children or early adolescence. Although PFAPA syndrome frequently occurs in pediatric patients, some published literatures have reported suspected cases in adults.3–8 However, it is important to note that monogenic autoinflammatory condition may either present in adulthood due to either de novo mutations or presence of mosaicism. Classic examples are cryopyrin-associated autoinflammatory syndromes and tumor necrosis factor receptor associated periodic syndrome.9,10 In addition, the phenotype of certain monogenic ones such as hyperimmunoglobulin D syndrome may be skewed in a growing child to become milder albeit requiring treatment based on persistent clinical symptoms and warranting a high index of suspicion from adult rheumatologists. 11 There are a growing number of multigenic autoinflammatory conditions that include Schnitzler’s syndrome; adult-onset Still’s disease (AOSD); synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome/ CRMO/chronic non-bacterial osteomyelitis (CNO); Adamantiades-Behçet’s disease; Yao syndrome (YAOS); and PFAPA syndrome.1,2,12,13 Among these conditions, patients with AOSD and Schnitzler’s syndrome share some common features like spiking fever and skin rash.12,14–16 Diagnosis and treatment of multigenic autoinflammatory diseases remain challenging due to their rarity and diverse presentations.
This review focuses on recent advances in genetically complex autoinflammatory diseases, including new insights regarding their pathogenesis, clinical overview, diagnosis, and management.
Methods
The articles included in this narrative review were identified through literature searches of English publications in the PubMed database using the MeSH terms or keywords: ‘Schnitzler’s syndrome’, ‘Still’s disease’, ‘AOSD’, ‘SAPHO syndrome’, ‘CRMO’, ‘Adamantiades-Behçet’s disease’, ‘YAO syndrome’, ‘PFAPA syndrome’, and search strings: ‘Schnitzler’s syndrome and pathogenesis’, ‘Schnitzler’s syndrome and biomarker’, ‘Schnitzler’s syndrome and diagnosis’, ‘Schnitzler’s syndrome and treatment’, ‘Schnitzler’s syndrome and IL-1 inhibitors’, ‘Schnitzler’s syndrome and canakinumab’. ‘AOSD and pathogenesis’, ‘AOSD and biomarker’, ‘AOSD and diagnosis’, ‘AOSD and treatment’, ‘AOSD and IL-1 inhibitors’, ‘AOSD and canakinumab’. ‘SAPHO syndrome and pathogenesis’, ‘SAPHO syndrome and biomarker’, ‘SAPHO syndrome and diagnosis’, ‘SAPHO syndrome and treatment’, ‘SAPHO syndrome and IL-1 inhibitors’, ‘SAPHO syndrome and canakinumab’. ‘CRMO and pathogenesis’, ‘CRMO and biomarker’, ‘CRMO and diagnosis’, ‘CRMO and treatment’, ‘CRMO and IL-1 inhibitors’, ‘CRMO and canakinumab’. ‘Adamantiades-Behçet’s disease and pathogenesis’, ‘Adamantiades-Behçet’s disease and biomarker’, ‘Adamantiades-Behçet’s disease and diagnosis’, ‘Adamantiades-Behçet’s disease and treatment’, ‘Adamantiades-Behçet’s disease and IL-1 inhibitors’, ‘Adamantiades-Behçet’s disease and canakinumab’. ‘YAO syndrome and pathogenesis’, ‘YAO syndrome and biomarker’, ‘YAO syndrome and diagnosis’, ‘YAO syndrome and treatment’, ‘YAO syndrome and IL-1 inhibitors’, ‘YAO syndrome and canakinumab’. ‘PFAPA syndrome and pathogenesis’, ‘PFAPA syndrome and biomarker’, ‘PFAPA syndrome and diagnosis’, ‘PFAPA syndrome and treatment’, ‘PFAPA syndrome and IL-1 inhibitors’, ‘PFAPA syndrome and canakinumab’. In all, 275 relevant articles were identified, reporting the results of studies in humans, published from inception to March 2021. The articles were scrutinized, and the references listed are relevant to the objective of this review.
Schnitzler’s syndrome
Overview
Schnitzler’s syndrome is a rare, acquired autoinflammatory disease, with chronic urticarial rash, lymphadenopathy, bone pain, fatigue, systemic inflammatory response along with monoclonal gammopathy as main features. 17 The exact prevalence of the disease is unknown but since 1972, fewer than 300 cases have been reported in the scientific literature.18–20
Pathogenesis
The etiology of Schnitzler’s syndrome is yet to be elucidated. Multiple features indicate that it is an acquired autoinflammatory disorder,21,22 with elevated levels of proinflammatory cytokine IL-1, including IL-1β, playing a central role in the pathophysiology of the disease.23,24 However, a possible link between autoinflammation and the monoclonal component (monoclonal IgM or IgG gammopathy) remains to be determined, because the level of monoclonal component is highly variable in Schnitzler’s syndrome, which can be very low or very high at once.25,26 Lipsker et al. performed immunoblotting on epidermal and dermal skin extracts, and immunoelectron microscopic studies on Lowicryl K4M-embedded skin sections in 3 patients with Schnitzler syndrome. Data showed that two patients had IgM deposits in the epidermis around the keratinocytes and below the lamina densa region of the anchoring fibrils, which suggest that monoclonal gammopathy plays a part in the pathophysiology of the skin rash and urticarial lesions. 27 Previous reports have shown that the aberrant nod-like receptor protein-3 (NLRP3) inflammasome signaling and cytokine pathway dysregulation play a key role in the pathogenesis of the disease.25,28 A study conducted in adult patients with Schnitzler’s syndrome has shown elevated levels of a chemoattractant, C-C motif chemokine ligand 2 [CCL2], in patients with Schnitzler’s syndrome, and there was a correlation between systemic CCL2 levels and the disease activity, suggesting the potential of CCL2 as a biomarker of disease activity and treatment response. 29 In a prospective study, ex vivo cytokine profiles of Peripheral Blood Mononuclear Cells (PBMCs) was evaluated in 36 adult patients with Schnitzler syndrome prior to treatment and after initiation of anti-IL-1 therapy. The spontaneous production of TNFα, IL-6, IL-1β, IL-1α, and IL-1RA by PBMCs was higher in patients with Schnitzler syndrome when compared with controls. In addition, the elevated levels of IL-1β, IL-1α, IL-6, and TNFα may represent an initial aspect of the cytokine signature in Schnitzler syndrome. 30 An additional finding on the T cell cytokine profile was decreased levels of Th1, Th2, and Th17, including reduced IL-10 levels. 30
Clinical manifestations and diagnosis
The main clinical features of Schnitzler’s syndrome include fever, musculoskeletal pain, lymphadenopathy, with chronic urticarial exanthema and immunoglobulin (Ig) monoclonal gammopathy (IgM or IgG) being the hallmarks of the disease.21,23 Patients with Schnitzler’s syndrome may develop lymphoproliferative disease 21 and, in rare cases, if the disease is not properly treated, amyloid A (AA) amyloidosis may occur. 23
Schnitzler’s syndrome is a diagnosis of exclusion due to its rarity, and it should be distinguished from other diseases which are more prevalent and have similar manifestations, 31 being chronic spontaneous urticaria the most crucial differential diagnosis. 23 The diagnosis of Schnitzler’s syndrome is based on the Lipsker’s criteria 32 or revised Lipsker’s criteria known as the Strasbourg’s criteria.18,23 The Strasbourg criteria for diagnosis (81% sensitivity for a clear diagnosis) of Schnitzler’s syndrome were defined at an international consensus meeting in 2012 and comprise two main and four secondary criteria (Figure 1). 23 These criteria differentiate patients with definite and probable Schnitzler’s syndrome. The skin biopsies of patients with Schnitzler’s syndrome have shown increase in expression of cytokines including IL-1β, IL-6, IL-18, myeloperoxidase (a marker of neutrophil activation), and inflammasome components, such as apoptosis-associated speck-like protein containing CARD (ASC) and caspase-1.23,33

Strasbourg criteria for the diagnosis of Schnitzler’s syndrome.
Treatment
Treatment with nonsteroidal antiinflammatory drugs (NSAIDs) and immunosuppressive drugs were less effective with inconsistent results. 32 IL-1 blockers have shown to be effective in the management Schnitzler’s syndrome.34,35 Data from a case report showed clinical improvement with the disappearance of fever, arthritis, and skin lesions in a patient with Schnitzler’s syndrome after treatment with the IL-1 receptor antagonist anakinra. 36 Rilonacept is an IL-1 trap molecule which can block IL-1α and IL-1β signaling. Data from a prospective study have shown that rilonacept was effective in patients with Schnitzler’s syndrome with significant reductions in health assessment scores and physician’s global assessment scores versus baseline levels. 37 In a retrospective study, out of 42 patients enrolled, 29 received anakinra and responded to the treatment. After a median follow-up of 36 months, the effectiveness of treatment remained unchanged. 35
Canakinumab, a fully human monoclonal antibody against IL-1β, was shown to induce a rapid and sustained clinical response and was effective in treating patients with Schnitzler’s syndrome. In a study, an adult patient with Schnitzler’s syndrome received canakinumab treatment every 8 weeks for 6 consecutive months and the clinical response to treatment was fast with sustained remission 38 (Table 1). Krause and colleagues 39 reported the results of a Phase II, randomized controlled study, which evaluated the effects of canakinumab in 20 adult patients with Schnitzler’s syndrome. Data showed that on Day 7, complete clinical response was significantly higher in patients who received canakinumab (n = 7) compared to patients who received placebo (n = 13) (71% vs 0%; p = .001). The long-term extension study of 15 patients with Schnitzler’s syndrome has shown that canakinumab could effectively reduce the clinical signs and symptoms of the disease, decrease the levels of inflammatory markers (CRP and SAA), and sustained the quality of life of the patients over 4 years. 28
Treatment options for the multigenic autoinflammatory diseases.

Approved by US FDA; #Approved by European Medicines Agency.
n, number of patients received the respective therapy.
Anakinra, canakinumab and rilonacept are IL-1 inhibitors; infliximab, etanercept and adalimumab are anti-TNF-α agents; tocilizumab and sarilumab are IL-6 inhibitors; secukinumab is an IL-17A inhibitor; NSAIDs, DMARDs, corticosteroids and glucocorticoids are conventional treatment.
Adult onset Still’s disease
Overview
AOSD is a rare, systemic autoinflammatory disorder characterized by a classic triad of high spiking fever, arthralgia/arthritis, skin rash, being commonly accompanied by hyperferritinemia, lymphadenopathy, and leukocytosis. The estimated incidence of AOSD is approximately 0.16 cases in 100,000 people in Western France,89,90 the annual incidence of AOSD is approximately 0.4 per 100,000 people in Northern Norway 91 and the estimated prevalence of AOSD is 3.9 per 100,000 people in Japan. 92 A study in patients with AOSD in Turkey reported an annual incidence of 0.62 and overall prevalence of 6.77 per 100,000 population. 93 Globally, the estimated annual incidence of AOSD is between 0.16 and 0.62 (per 100,000 persons) and its approximate prevalence 1–24 per million.90,91,94,95 Data from US nationwide inpatient sample (NIS) database have shown that 5,820 AOSD patients were hospitalized between 2009 and 2013. 96 AOSD is associated with significant morbidity-mortality and life-threatening manifestations, with a negative impact on the health-related quality of life of patients. 96 Macrophage activation syndrome is a severe, life-threatening complication of both sJIA and AOSD.97–100
Pathogenesis
The pathogenesis of AOSD is complex and not clearly understood. Studies have shown that activation of the innate immune system and dysregulation of proinflammatory cytokines together mediate the pathogenesis of AOSD. 101 Increased levels of NLRP3 inflammasome and its correlation with disease activity suggests the involvement of NLRP3 in the pathogenesis of AOSD. 102 The activation of inflammasome NLRP3 can lead to overproduction of cytokines IL-1β and IL-18.102–105 These cytokines can trigger the dysregulation of downstream inflammatory cytokines, such as IL-6, IL-8, IL-17, and TNF-α, which are involved in the pathogenesis of AOSD.15,105–110 Levels of the serological marker serum ferritin are often elevated in patients with AOSD, with ferritin elevations correlating with disease activity. Serum ferritin levels > 5 times the normal upper limit in fever of unknown origin suggest AOSD with a sensitivity of up to 80%, which can increase to 93% if combined with glycosylated ferritin of less than 20%. 89 In addition, there is increasing evidence of uncontrolled T-cell and macrophage activation – inducing proinflammatory cytokines, such as IL-1, IL-6, IL-18, TNF-α, and interferon (IFN)-γ overproduction – playing a crucial role in the pathogenesis of AOSD.15,51,106,111
A study has shown higher levels of serum IL-33 and its soluble ST2 receptor in patients with active AOSD. 112 With reduction of disease activity, there is a decrease in serum IL-33 levels. Therefore, the IL-33/ST2 signaling pathway might play a key role in the pathogenesis that causes inflammation and skin manifestations in AOSD. 112 Another study which evaluated SDF-1/CXCL12 and soluble CXCR4 (sCXCR4) levels and their clinical relevance observed higher levels of serum CXCL12 and serum sCXCR4 in patients with AOSD versus healthy volunteers. The results from this study showed that sCXCR4 is a potential clinical biomarker for AOSD and higher levels of CXCR4/CXCL12 may trigger the inflammatory conditions and skin manifestations associated with AOSD. 113 Higher levels of IL-18 have been identified in AOSD patients.97,111,114,115
Clinical manifestations and diagnosis
Patients with AOSD usually present with high spiking quotidian fever, arthralgia and/or arthritis, a characteristic salmon-colored skin rash (usually concomitant with episodes of fever), lymphadenopathy, leukocytosis, and hyperferritinemia.12,14–16 A study investigated 66 patients with AOSD over an 18-year period and most prominent symptoms observed in patients were high fever (91%), arthralgia (89%) and cutaneous rash (56%). 116 Joint involvement is common in patients with AOSD; arthritis, usually mild and localized at the beginning, can aggravate through the course of the disease, becoming more severe and polyarticular. The most commonly involved joints are the knees, wrists, ankles, elbows, and proximal interphalangeal joints. AOSD often spares the distal interphalangeal joints of the hand and shoulders. Narrowing of the intercarpal or carpometacarpal joint spaces has been observed in up to 75% of patients with chronic articular AOSD.43,116–121 The characteristic rash in Still’s disease is transient, nonpruritic, salmon-colored, with macular or maculopapular lesions, which are often observed during febrile episodes, predominantly in the late afternoon or evening. The typical rash appears mainly on the trunk and proximal extremities, in some occasions being misdiagnosed as an adverse reaction to medicines.116,122 In rare cases, neurological and vascular manifestations are present, and the diagnosis may become more challenging due to a less frequent clinical presentation. 123 A rapid and efficient, non-invasive diagnosis method is needed for a better 43 management of AOSD. 124
The diagnosis of AOSD depends on the disease presentation upon exclusion of differential diagnoses including infection, malignancy, and other rheumatic inflammatory diseases. 125 Although AOSD has been regarded as being part of the same disease spectrum of systemic juvenile idiopathic arthritis (sJIA), the patterns of disease course, their clinical and laboratory features may differ between adults and children, possibly because children – with a more naïve immune system – may react differently on first exposure to antigens. The current ILAR classification criteria for sJIA are based on clinical and analytical manifestations in the first 6 months of illness and the presence of arthritis at presentation is mandatory. On the other hand, the Yamaguchi criteria for AOSD diagnosis, only require the presence of arthralgia for more than 2 weeks. 126 Comparison of clinical characteristics of AOSD and sJIA have shown significant female predominance among patients with AOSD (p < 0.05). Significantly increased serum ferritin levels, hepatomegaly, and splenomegaly were observed more frequently with AOSD (p < 0.05). Arthritis was more common among sJIA patients than among AOSD patients (p < 0.05).97,127 More AOSD patients versus sJIA patients presented with a sore throat (81% vs 46%; p = 0.03). 128 AOSD patients had a significantly higher serum ferritin concentration than sJIA patients during the active disease phase (p < 0.01).127,128
To identify AOSD patients, the Yamaguchi’s criteria are widely used being a sensitive tool (sensitivity 96.2% and specificity 92.1%) for diagnosis and classification of AOSD 129 (Table 2), followed by Cush’s criteria (sensitivity 80.6%)130–132 and Fautrel’s criteria (sensitivity 80.6% and specificity 98.5%) (Table 2).133–135 Recently, Crispín’s clinical scale (sensitivity 76.9% and specificity 98%) (Table 2) has also been adopted.89,135
Criteria for diagnosis of AOSD.

Five or more criteria are required, with two or more being major criteria for diagnosis of AOSD.
Four or more major criteria are required, or three major and two minor criteria.
At least 30 points in patients with fever of unknown origin allows for the diagnosis of AOSD without further studies (specificity ~98%).
ALT, alanine aminotransferase; ANA, antinuclear antibody; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; PMN, polymorphonuclear; RF, rheumatoid factor
[Source: Seco et al. 89 ].
Treatment
NSAIDs, disease-modifying anti-rheumatic drugs (DMARDs), glucocorticoids or corticosteroids have been used as conventional treatments for AOSD.40–44,51,136 Tocilizumab, an IL-6 inhibitor, has shown efficacy in the treatment of patients with refractory AOSD.50–52,137,138 Data from a multicenter retrospective study involving 27 patients ⩾ 16 years of age with AOSD showed that the remission was achieved in all patients, except one, with either anakinra or tocilizumab treatment. Response to treatment mainly depended on disease phenotype, such as arthritis and a chronic articular phenotype was associated with response to tocilizumab therapy, whereas the systemic form and the absence of arthritis were associated with response to anakinra therapy. This study has shown the therapeutic implications of the phenotypic dichotomy of AOSD. 94 This suggests that AOSD treatment can be tailored, at least in some patients, based on the clinical presentation. There are limited data on the effectiveness of rilonacept in AOSD, but available reports have shown that rilonacept is effective in reducing the symptoms related to arthritis and other systemic symptoms, in patients with AOSD.15,139,140
Canakinumab has shown rapid and sustained improvement of both articular and systemic features in AOSD. 49 Canakinumab was approved by the US Food and Drug Administration and the European Medicines Agency for Still’s disease, which is supported by the concept of a disease continuum that includes both SJIA and AOSD. 141 Feist et al. 46 have shown that treatment with canakinumab in a subgroup of patients with sJIA ⩾ 16 years of age, who are representative of AOSD patients, led to an improvement in the systemic and arthritic components of the disease in older adolescents and young adults. The available data indicate that canakinumab is effective and well tolerated for treating AOSD.45,47–49,110,142,143 A study has shown that treatment of adult patients with AOSD and active joint involvement with canakinumab led to a clinically significant improvement of outcome measures including DAS28-ESR, DAS28-CRP, ACR30, ACR50 and ACR70 and remission, compared with placebo. It was difficult to complete the recruitment of this trial due to the rarity and severity of the disease as well as the conditional approval of canakinumab for AOSD by the EMA; therefore, the study was terminated prematurely. 47 A retrospective real-life study in a cohort of 50 adult patients with refractory Still’s disease have shown high rates of sustained remission in patients treated with canakinumab. A complete response was observed in 78% of patients. Of 50 patients, 37 had a follow-up longer than 1 year and 27 more than 2 years and canakinumab had a long-term efficacy in the majority of patients with a significant steroid sparing effect. 144
SAPHO/CRMsO/CNO
Overview
SAPHO syndrome is a rare disease with chronic osteoarticular and dermatological disorders.145,146 SAPHO generally occurs in adults. 146 SAPHO affects the skeletal system including the anterior chest wall, long bones and spine, and may involve polyostotic lesions, as seen in bone scintigraphy the characteristic “bull’s head”, which is a sign of increase tracer uptake in the regions of sternocostoclavicular and manubriosternal joints.146,147 The annual prevalence of SAPHO syndrome varies from 0.00144 in 100,000 people in Japan to less than 1 in 10,000 in the White population.148–150 In another report, the annual prevalence of SAPHO is estimated to be less than 1/10,000.151,152 In a cross-sectional study on a cohort of SAPHO patients, prevalence of fibromyalgia in SAPHO syndrome was approximately 18%. 151
CNO/CRMO are rare diseases with disorders of chronic osteoarticular inflammation and skin manifestations. 146 CRMO/CNO predominantly affects children.146,153 CRMO/CNO affect the skeletal system including the anterior chest wall, long bones and spine, and may involve polyostotic lesions. 146 The estimated prevalence of CRMO/CNO is 1–2 per million population, mostly affecting children. 154 Incidence rate of CNO was estimated at 0.4 per 100,000 children/year in Germany.155,156 A chart review from a retrospective single-center analysis reported incidences of CNO increased from approximately 2.5 cases per year (1998–2007) to 4.7 CNO cases per year, which suggest that the suspected lower incidence rate of CNO may be an underestimation. 155
Pathogenesis
The precise pathogenesis of SAPHO syndrome remains unclear. However, it is believed to be related to genetic factors, infections, and immune dysregulation. 157 As in Schnitzler’s syndrome and AOSD, overproduction of IL-1β was also reported in SAPHO syndrome. 158 Studies have shown dysregulation of cytokines, including IL-1β, IL-8, IL-17, IL-18, and TNF-α, to be potentially involved in the pathogenesis of SAPHO.159,160 A study that measured the expression of receptor activator nuclear factor κ-Β ligand (RANKL)/osteoprotegerin (OPG) in patients with SAPHO syndrome reported higher levels of RANKL in patients with active disease.150,161 Another study, comparing patients with SAPHO with healthy controls, found that the proportion of natural killer (NK) cells was significantly lower in SAPHO patients and the ratio of T-helper cell 17 (Th17) and regulatory T-cells (Treg) was higher. A depletion of NK cells and an imbalance of Th17 and Treg cells in SAPHO patients could be related to immune inflammation-mediated pathogenesis. 162
JAK/STAT signaling pathway may play a role in the pathogenesis of SAPHO syndrome. A retrospective review of data from a cohort of SAPHO patients has shown benefits with the use of tofacitinib, a Janus kinase (JAK) 1/3 inhibitor. A decrease in systemic inflammation, reduction in pain and rash, improvement in quality of life and remission on MRI was observed with tofacitinib treatment. 163 Although tofacitinib is not approved for use for SAPHO, these findings provide evidence that tofacitinib could be a potential therapeutic option for the treatment of SAPHO syndrome.163,164
Studies have shown that activation and expression of inflammasome components, such as apoptosis-associated speck-like protein (ASC), NLRP3, and caspase-1, are elevated in CRMO/CNO patients, which may contribute to pathogenesis of the disease.154,165,166
Clinical manifestations and diagnosis
Patients with SAPHO syndrome present with sterile joint inflammation (synovitis), skin manifestations, such as severe acne and palmoplantar pustulosis, hyperostosis and non-infectious osteitis.146,167,168 In CRMO/CNO, symptoms are heterogeneous, such as swelling of the affected area, severe bone pain, fever, malaise, and weight loss. The symptoms related to bone inflammation and pain can often get worse at night. In patients with CRMO/CNO, sporadic inflammatory bone pain occurs as recurrent flares due to osteomyelitis, which presents as multiple aseptic foci. 154
Diagnosis of SAPHO/CRMO/CNO is based on clinical and radiological findings. 146 The diagnosis of CRMO/CNO is mainly achieved by exclusion of differential diagnoses, such as bacterial osteomyelitis and malignancy.154,169,170
For diagnosis of patients with CNO, clinical score from Jansson et al., 171 blood test, bone biopsy, microbiological investigations, radiological findings on MRI are generally used. 155 The Jansson score includes white blood cell counts, symmetric bone lesions, marginal sclerosis, normal body temperature, vertebral, clavicular, or sternal lesions, greater than two lesions in radiological imaging, and CRP ⩾ 10 mg/l. Scores of 39 or higher indicate CNO. 155 The clinical presentation of CNO range from self-limited unifocal to chronic recurrent multifocal bone inflammation.155,165,172,173
In most SAPHO/CRMO/CNO patients, polyostotic involvement is observed. Bone scintigraphy has shown a characteristic ‘bull’s head’ sign in the sternocostoclavicular and manubriosternal joints, which is considered as a characteristic of SAPHO syndrome.146,174,175 The most widely applied diagnostic criteria for SAPHO syndrome were developed by Benhamou and colleagues176–178 (Table 3).
Diagnostic criteria for SAPHO syndrome.
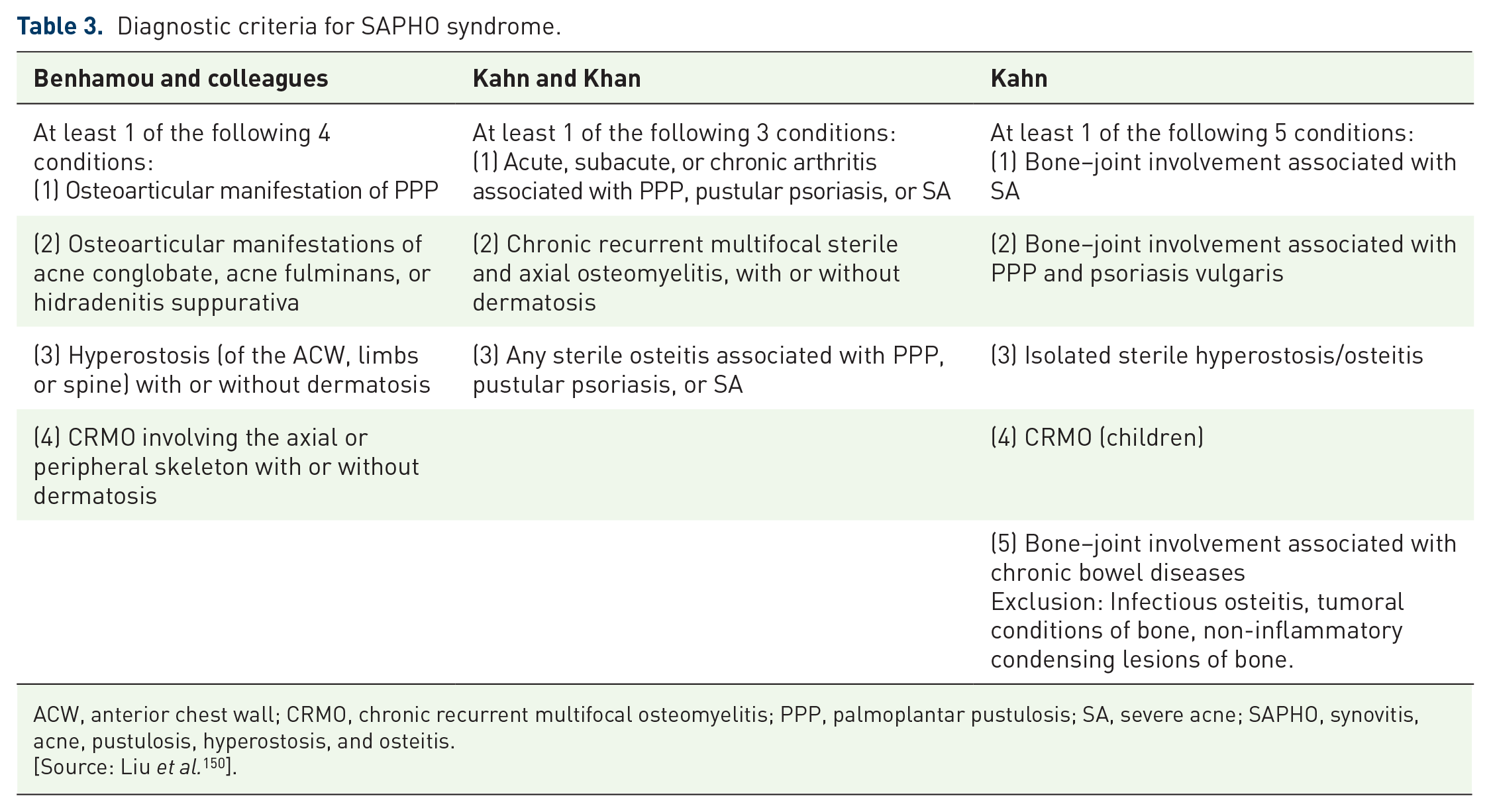
ACW, anterior chest wall; CRMO, chronic recurrent multifocal osteomyelitis; PPP, palmoplantar pustulosis; SA, severe acne; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis.
[Source: Liu et al. 150 ].
Treatment
Conventional treatments for SAPHO include NSAIDs, DMARDs, glucocorticoids or corticosteroids, bisphosphonates and antibiotics.157,167,179 NSAIDs, DMARDs, glucocorticoids or corticosteroid, bisphosphonates and antibiotics have also been used as conventional treatments for CRMO/CNO.180–182 Pamidronate, a bisphosphonate, has shown efficacy on osteoarticular manifestations of SAPHO, but did not improve cutaneous lesions.157,183 Gil et al. 184 described a case in which an adult patient with SAPHO syndrome who developed palmoplantar pustulosis and sternoclavicular osteitis received treatment with bisphosphonate, systemic corticosteroid, and cyclosporine which resulted in complete resolution of the articular and dermatologic manifestations without any side effects. Bisphosphonate has shown a favorable treatment response in pediatric SAPHO syndrome. 185 In an observational study, clinical data of 24 adult patients with SAPHO syndrome were analyzed. Out of 17 patients who received the combination treatments of NSAIDs and DMARDs, 15 experienced an improvement in their symptoms. In total, 18 patients with SAPHO syndrome received bisphosphonates, and 4 patients received TNF blockers. Among the patients who received TNF blockers, adalimumab was ineffective in one patient; however, the disease activity was controlled in that patient by add-on treatment with DMARDs. Patients with SAPHO syndrome responded to NSAIDs, DMARDs, and bisphosphonates combination therapy. TNF blockers were shown to be effective in patient with SAPHO syndrome refractory to NSAIDs and DMARDs. 186 The CNO/CRMO subgroup of the Childhood Arthritis and Rheumatology Research Alliance (CARRA) held meetings to develop standardized treatment regimens for CNO/CRMO and enable comparative effectiveness treatment strategies, which resulted in 3 consensus treatment plans for pediatric patients with CNO/CRMO refractory to NSAIDs. The 3 consensus treatment plans are sulfasalazine or methotrexate, TNF inhibitors with optional methotrexate, and bisphosphonates. 187
TNF-α antagonists have shown a favorable treatment response in pediatric SAPHO syndrome. 185 TNF inhibitors, such as etanercept, adalimumab, infliximab are generally used in SAPHO syndrome after methotrexate failure. 54 Anti-TNF-α agents have shown promising results in SAPHO syndrome167,179,185,188 and in CRMO/CNO.55,172,182,189 In a case study, tocilizumab was shown to be effective in controlling symptoms and inflammatory markers in adult patients with CRMO/CNO. 56
IL-inhibitors also seem to be effective in the treatment of patients with SAPHO and CRMO/CNO. As per available data related to IL-1 inhibition, most patients with SAPHO show a significant improvement in musculoskeletal manifestations. 188 IL-1 inhibition with anakinra has shown efficacy in the treatment of SAPHO syndrome. In a case study, anakinra treatment in a patient with SAPHO syndrome resulted in resolution of systemic and osteoarticular symptoms.58,159 In a study, treatment with canakinumab was associated with variable outcomes in patients with CRMO/CNO. 60 Secukinumab, an IL-17A inhibitor, successfully treated refractory mandibular lesions in an adult patient with SAPHO syndrome. 61
Adamantiades-Behçet’s disease
Overview
Adamantiades-Behçet’s disease is a multisystem, autoinflammatory disease of unknown etiology characterized by recurrent painful mucocutaneous ulcerations, and idiopathic systemic vasculitis that affects both large and small blood vessels. It can also present with arthralgia/arthritis, other skin lesions uveitis and neurological manifestations.190–198 The incidence of Adamantiades-Behçet’s disease varies according to geographic location, with the highest prevalence of 20–420 (per 100,000 people) in Turkey, followed by 80 in Iran, 20 in Saudi Arabia, and 17 in Iraq. 199 In the United Kingdom, the prevalence of Adamantiades-Behçet’s disease is 0.64 cases per 100,000 population.199,200 A study in United States reported an overall incidence of 0.38 per 100,000 population and prevalence rate of 5.2 per 100,000 population. 201
Pathogenesis
The pathogenesis of Adamantiades-Behçet’s disease is still unclear. IL-1 seems to play a crucial role in the pathogenesis of Adamantiades-Behçet’s disease, as IL-1 levels were shown to be significantly higher in patients with Adamantiades-Behçet’s disease versus healthy controls.202–204 Elevated levels of serum IL-6 were also seen in patients with active Adamantiades-Behçet’s disease, which may correlate with disease activity.205–207
Functional abnormalities of Toll-like receptors in monocytes have been noted in patients with Adamantiades-Behçet’s disease, and may be correlated with disease activity. One of the most consistent findings in the pathogenesis of Adamantiades-Behçet’s disease is hyperactive state of neutrophils. The antigen presenting cells (macrophages) stimulate immune cells by Toll-like receptors activation and the innate and adaptive immune pathways together with neutrophil activation play a crucial role in the pathogenesis of Adamantiades-Behçet’s disease.195,208
Histocompatibility leucocyte antigen (HLA)-B51, a class I MHC antigen, represents the strongest genetic risk factor associated with Adamantiades-Behçet’s disease. Other HLA-independent loci, such as IL23R/IL12RB2 and IL10 (allele rs1518111 A) have also been associated with Adamantiades-Behçet’s disease.195,209 Data from a gene expression study, which identified genes that might be related to pathogenesis of Adamantiades-Behçet’s disease, have shown that 1103 genes and 652 genes were differentially expressed in the CD8 + CD27−CD28− and CD8 + CD27 + CD28 + T-cell subsets, respectively, of patients with Adamantiades-Behçet’s disease. 210 A case-control study found that the gene expression of TNF-α increased in patients with Adamantiades-Behçet’s disease, suggesting that TNF-α has a role in the pathogenesis of the Adamantiades-Behçet’s disease. 211
Clinical manifestations and diagnosis
The notable clinical manifestations in patients with Adamantiades-Behçet’s disease are recurrent oral and genital ulcerations, and ocular inflammation. Articular, neurological, and gastrointestinal involvement are also reported.190–193 Cardiopulmonary signs and symptoms may be the first manifestation. 197 The diagnosis of Adamantiades-Behçet’s disease is challenging, because it is mainly based on clinical manifestations, which are frequent manifestations (oral aphthosis, genital aphthosis, pseudofolliculitis, erythema nodosum) also seen in other diseases. 199 The International Criteria for Behçet Disease (ICBD) was developed in collaboration with the experts from 27 countries to address the diagnostic dilemma of Adamantiades-Behçet’s disease. ICBD is a point score system, a scoring ⩾ 4 is classified as Adamantiades-Behçet’s disease. As per ICBD, ocular lesions, genital aphthosis and oral aphthosis are assigned 2 points each, while skin lesions, vascular manifestations and neurological manifestations are assigned 1 point each. If pathergy test is done, an additional 1 point is assigned. 212 The diagnostic criteria for Adamantiades-Behçet’s disease are presented in Table 4. 212
International criteria for Adamantiades-Behçet’s disease. 212


Treatment
NSAIDs, DMARDs, glucocorticoids or corticosteroids have been used as conventional treatments for Adamantiades-Behçet’s disease.62–66,213 Higher doses of corticosteroids are effective for treatment of acute exacerbations of Adamantiades-Behçet’s disease with vascular involvement, NBD and refractory or severe gastrointestinal involvement.198,214 The treatment goal is to prevent irreversible organ damage by suppressing inflammatory exacerbations. 198 A combination of immunosuppressants, such as azathioprine, cyclosporine-A or cyclophosphamide is recommended for the management of acute deep vein thrombosis in Adamantiades-Behçet’s disease.198,214 Oral and genital ulcers should be treated with topical steroids. Apremilast, an oral phosphodiesterase 4 inhibitor, is effective for oral ulcer, which is the most common symptom of Adamantiades-Behçet’s disease. 215 Apremilast has been approved for treatment of recurrent oral ulcers in patients with Adamantiades-Behçet’s disease in Japan after successful clinical trials.216,217 Studies have shown that colchicine was effective in preventing acute arthritis, recurrent mucocutaneous lesions and arthritis episodes of Adamantiades-Behçet’s disease. In a 2-year randomized, controlled study, 116 adult patients with Adamantiades-Behçet’s disease with active mucocutaneous disease without eye or major organ involvement, received either colchicine or placebo. Data showed that compared to placebo group, complete response (sustained absence of any lesions) to treatment was significantly higher in colchicine group, which showed reduced occurrence of genital ulcers (p = 0.004), erythema nodosum (p = 0.004), and arthritis (p = 0.033) in women, and reduced occurrence of arthritis (p = 0.012) in men. Colchicine was effective for treating the arthritis and some of the mucocutaneous manifestations of Adamantiades-Behçet’s disease.198,214,218,219
Emerging evidence supports the use of biologics in patients with refractory, severe Adamantiades-Behçet’s disease, and especially for those with ocular,220,221 central nervous system, 221 and gastrointestinal70,221,222 involvements, anti-TNF-α therapy has been shown to have benefits.68,69,198 Infliximab and adalimumab can be useful in managing patients with intestinal Adamantiades-Behçet’s disease, especially in severe or refractory cases. 223 In a case study, tocilizumab has been shown to be effective in the treatment of intestinal Adamantiades-Behçet’s disease. 224
Anakinra and canakinumab have shown some benefit in Adamantiades-Behçet’s disease including in patients with mucocutaneous manifestations and in intestinal Adamantiades-Behçet’s disease.71,74,214,223,225–228 Data from studies have shown that IL-1 inhibitors could effectively control mucocutaneous and ocular manifestations in patients with Adamantiades-Behçet’s disease.229–231 In a retrospective study, 36 patients with Adamantiades-Behçet’s disease treated with anakinra or canakinumab were enrolled and divided into two groups. Patients showing a treatment duration of at least 52 weeks and no secondary inefficacy during the first follow-up year were included in group 1 and patients showing primary or secondary inefficacy within the first 52 weeks of treatment were included in group 2. Anakinra was administered in 13/18 patients and canakinumab in 5/18 patients, in both the groups. Data from this study showed that within the 3-month follow-up, clinical response (complete resolution of all active Adamantiades-Behçet’s disease-related clinical manifestations) was obtained in 100% (18/18) of patients in group 1% and 38.9% (7/18) of patients in group 2 (p < 0.0001), which demonstrated that anakinra and canakinumab were effective in Adamantiades-Behçet’s disease. 232 In another retrospective study, 19 patients with Adamantiades-Behçet’s disease-uveitis received treatment with IL-1 inhibitors (anakinra [n = 13] and canakinumab [n = 6] and followed-up for 12 months. The number of patients presenting with ocular flares (ocular inflammatory manifestations that occurred after a period of remission) and the number of reported ocular flares decreased during the 12-month follow-up period. Anakinra and canakinumab were shown to be effective in managing Adamantiades-Behçet’s disease-related uveitis with a significant reduction of ocular inflammatory flares. 233
Rituximab, alemtuzumab, and ustekinumab have been evaluated in a number of studies and were shown to be effective in Adamantiades-Behçet’s disease. Sadreddini et al. reported one case in which a patient with Adamantiades-Behçet’s disease with visual loss due to retinal vasculitis was resistant to prednisolone and azathioprine treatment, was successfully treated with rituximab and disease remission was sustained for 24 months. In another study, 32 patients with Adamantiades-Behçet’s disease received alemtuzumab and 84% patients achieved partial or complete remission after receiving the first course of alemtuzumab. Relapse-free survival rates was observed in 83.6% patients at 6 months. Another case study reported an improvement in PASI 50 and PASI 75 scores in patients with Adamantiades-Behçet’s disease treated with ustekinumab.76–78,234–237 In an open-label prospective study, 30 patients with Adamantiades-Behçet’s disease, who were diagnosed with active oral ulcers resistant to colchicine, received ustekinumab. Compared with baseline, the median number of oral ulcers per patient was significantly lower (p < 0.0001) at Week 12 with ustekinumab treatment. Complete response was achieved in 60.0% of patients at Week 12% and 88.9% of patients at Week 24. Ustekinumab was effective in treating Adamantiades-Behçet’s disease related oral ulcers that are resistant to colchicine treatment. 238
Yao syndrome
Overview
YAOS, formerly called nucleotide-binding oligomerization domain 2 (NOD2)-associated autoinflammatory disease, is characterized by fever, polyarthritis, dermatitis, distal extremity swelling, gastrointestinal and sicca-like symptoms associated with specific variants of NOD2 sequence. YAOS is predominantly observed in white adults at an estimated disease prevalence of 1–10 per 100,000 population. 239 It might be the most common form of sporadic autoinflammatory disease occurring in adults.22,239,240
Pathogenesis
The molecular mechanisms and underlying pathogenesis of YAOS are unclear, but there is a genetic association with certain NOD2 variants.239,241 A study that examined NOD2 expression and activation of signaling pathway reported that NOD2 transcript level and basal p38 mitogen-activated protein kinase activity was elevated in peripheral blood mononuclear cells from IVS8 + 158 YAOS patients. Also, the cells of the patients had high basal IL-6 levels which were increased by muramyl dipeptide stimulation, indicating that NOD2 expression and pathway activation are aberrant in YAOS. 242 In a case series, 241 genetic testing in three YAOs patients detected presence of the following heterozygous NOD2 variants:
Q902 K/rs201035873 (c.2704 C > A) in exon 7
R541 P/rs753857915 (c.1622G > C) in exon 4
Y514 H/rs540122692 (c.1540 T > C) in exon 4
A recent study 243 reported that the following NOD2 variants are linked to YAOS:
c.2555A > G (p. Asn852Ser) in exon 6
c.2264 C > T (p. Ala755Val) in exon 4
c.2774A > G (p.asp925gly) in exon 8
c.160G > A (p. Glu54Lys) in exon 2
c.140 C > T (p.S47L) in exon 2
IL-1 antagonist can improve the overall symptoms and physical function in patients, which suggest a potential role of IL-1 in the pathogenesis of YAOS. 79
Clinical manifestations and diagnosis
As in AOSD, recurrent fever may occur in patients with YAOS, with febrile episodes lasting from days to several weeks. Erythematous patches or plaques can be seen in patients with YAOS, and recurrent mild to moderate abdominal pain, bloating, cramping, and diarrhea may also be present.80,240,241 YAOS is a systemic inflammatory disease which can lead to chronic pain syndrome and disability, but it rarely affects the solid internal organs. 79
The diagnosis of YAOS is mainly dependent on the phenotype and genotype characteristic of the disease.22,79,244 The criteria for diagnosis of YAOS are summarized in Table 5.79,240 Endoscopic, radiological, or histologic examinations have shown no evidence of inflammatory bowel disease. There may be an occasional non-specific focal colitis away from the ileum. Arthritis can occur periodically in approximately 90% of patients. Asthma and urinary stones are common comorbidities associated with YAOS. 240 Early recognition of the disease can minimize extensive testing, and with a correct diagnosis, physicians will be able to better manage the disease.22,79
Diagnostic criteria for Yao syndrome.

Yao syndrome is diagnosed if two major criteria, at least one minor criterion, the molecular criterion, and exclusion criteria are fulfilled.
The genetic testing was performed at the Center for Genetic Testing at Saint Francis, Tulsa, Oklahoma.
[Source: Yao and Shen 79 ]
Treatment
The therapeutic regimen for YAOS consists of glucocorticoids and/or sulfasalazine as the first-line treatment option. Glucocorticoids have been shown to reduce disease severity and duration of flares, and treatment with sulfasalazine resulted in a significant symptomatic improvement in patients with YAOS.22,79,240
In a retrospective study, tocilizumab was shown to improve the symptoms and physical function of adult patient with YAOS. 79 Treatment with canakinumab was also shown to be effective, which improved the overall symptoms and physical function of the patients with YAOS.22,79,240 Data from a retrospective study have shown that canakinumab was effective and well tolerated in adult patients with YAOS and may be considered as a therapeutic option. 80
PFAPA syndrome
Overview
PFAPA syndrome is the most common autoinflammatory disease in children, but its hereditary basis remains uncharacterized and specific genetic causes are yet to be identified.245,246 Although, PFAPA is the most common periodic fever disease in children, some studies have reported it in adults.3,4 PFAPA syndrome has a delayed onset during adulthood. Clinical features of PFAPA in adults which are mainly overlapping with those of children are fever, oral aphthous, pharyngitis, cervical lymphadenopathy, abdominal pain, headache, and nausea. 247 Among the key signs and symptoms of PFAPA syndrome, the occurrence of recurrent fever with erythematous pharyngitis represent the most strongly associated diagnosis of PFAPA syndrome in adulthood. 248 The estimated incidence of PFAPA is 2.3 per 10 000 children up to 5 years of age in Norway.1,249
Pathogenesis
PFAPA is a genetically complex autoinflammatory disease with disorders of innate and adaptive immunity. 250 PFAPA syndrome indicates a heterogenous, polygenic, or complex inheritance. In several PFAPA cohorts, two types of mutations with a functional effect on the inflammasomes (MEFV E148Q and NLRP3 Q703 K) were seen. Therefore, inflammasome-related genes may play a role in PFAPA pathogenesis. 251 The exact pathogenetic processes underlying the symptoms of PFAPA syndrome are still unknown. An analysis of the upstream regulator of immune pathways indicated that activation of toll-like receptors (TLRs), production of IFN-γ, and decrease in 1,25-dihydroxyvitamin D may be associated with the immune pathways involved in the pathogenesis of PFAPA. 252 The elevated serum IL-1β concentrations and elevated proinflammatory cytokine production by monocytes of PFAPA patients upon stimulation of lipopolysaccharide indicate that dysregulation of the immune responses could be associated with PFAPA flares.252–254 Previous reports have shown that a variant upstream of IL-12A (rs17753641) is associated with PFAPA syndrome. The monocytes from patients heterozygous or homozygous for the rs17753641 risk allele secreted higher levels of IL-12p70 upon stimulation with IFN-γ and lipopolysaccharide. Data also suggest that abnormal antigen-presenting cell function, and T-cell activity and polarization, are associated with the pathogenesis of PFAPA. 255 An analysis in patients with PFAPA showed that B-cell adapter protein (PIK3AP1) and spondin-2 (SPON2) gene regions are differentially methylated, which suggests that PIK3AP1 and SPON2 could play a role in PFAPA syndrome. 246 There are few studies which have surveyed familial clustering in PFAPA syndrome, and some reports from those studies are in favor of positive family history, which have shown that siblings and parents of PFAPA patients are similarly affected.245,256–258 In a study, comprehensive genetic analysis of families with PFAPA syndrome has shown the absence of a common gene harboring exonic mutations in the affected patients, which indicates that PFAPA syndrome is either inherited as a Mendelian disease with high genetic heterogeneity, or transmitted as an oligogenic or complex trait. 259 One study reported 27% of variants in MEFV and NLRP3 genes in a cohort of 62 patients with PFAPA. 260 In another study two mutations were observed, one in NLRP3 and one in MEFV, both known to be associated with PFAPA and found in one family. 259 In a study by Padeh et al., 28 patients of Arab and Jewish origin with PFAPA syndrome were analyzed of which six patients (21%) were found heterozygotes for M694 V and were carriers of FMF gene. The authors indicated that the reported frequency of FMF genes might be similar to that of a normal population. 261
Clinical manifestations and diagnosis
PFAPA syndrome is characterized by recurrent fever, as in AOSD and YAOS, which last for approximately 3–6 days and occurring once every 3–8 weeks. Other characteristics – which include pharyngitis, stomatitis, cervical adenitis, and leukocytosis – are variable in their appearance.246,262,263 As per recent reports, there are genetic similarities between PFAPA and Behçet’s syndrome. PFAPA shares risk loci at IL12A, STAT4, IL10, and CCR1-CCR3 with Behçet’s syndrome and recurrent aphthous stomatitis.264,265 Based on some common genetic and clinical similarities, it has been suggested that PFAPA together with recurrent aphthous stomatitis and Behçet’s syndrome form a construct which Manthiram et al. proposed to name Behcet Spectrum of Disorders.264,265
PFAPA syndrome is diagnosed based on clinical criteria and by exclusion of other etiologies. Elevated white blood cell counts and C-reactive protein levels may be seen during episodes. 266 Differential diagnoses include infectious pharyngitis and cyclic neutropenia. The absence of symptoms related to seasonal flares, upper respiratory tract infection, and negative pharyngeal swabs can facilitate diagnosis. The main differential diagnosis depends on the age of patients. Compared with children, the clinical picture is more heterogeneous in nature in adults; hence, PFAPA can be a diagnostic challenge for treating physicians.81,248 For PFAPA, as per Eurofever/PRINTO clinical classification, at least 7 criteria need to be met out of 8 proposed criteria, which are presence of pharyngotonsillitis, cervical lymphadenitis, periodicity and duration of episodes 3–6 days, and absence of diarrhea, chest pain, skin rash, and arthritis. 267 PFAPA diagnostic criteria according to Marshal, Pedeh and Cantarini are presented in Table 6.81,248,261,268
Diagnostic criteria for PFAPA according to Marshal, Padeh and Cantarini.

[Source: Więsik-Szewczyk et al. 81 ].
Treatment
The pharmacological management of PFAPA includes colchicine for prophylaxis of attacks. Data from two randomized controlled trials which compared tonsillectomy against non-surgical interventions have shown the effectiveness of tonsillectomy in children with PFAPA syndrome. 269 Tonsillectomy with or without adenoidectomy can be useful in some pediatric patients,81,269 whereas available data suggest that tonsillectomy is not so effective in adults.81,247 At the onset of fever in patients, prednisone can be effective in the resolution of fever episodes in pediatric 81 and adult patients. 86 Corticosteroids are effective for the resolution of flares in pediatric270–272 and adult81,270,272 patients with PFAPA. Steroids can be used during each episode. 1 Prednisone use has shown improved response in adult patients.81,247 The use of prednisolone at the time of fever onset has been shown to be useful in a randomized clinical trial because fever was decreased in pediatric patients with PFAPA syndrome. 273 Studies have shown that colchicine might be an effective preventive therapy in pediatric patients with PFAPA.84,261,274 Pediatric85,88,275 and adult86,87,275 patients with PFAPA have shown a prompt clinical response to the IL-1 inhibitors, anakinra and canakinumab.
Conclusion
Schnitzler’s syndrome, AOSD, SAPHO syndrome/ CRMO/CNO, Adamantiades-Behçet’s disease, YAOS, and PFAPA syndrome are a group of rare multigenic autoinflammatory diseases that can present varied clinical features. Diagnosis of these diseases can be challenging due to a combination of nonspecific manifestations that can also be seen in a variety of other conditions. Diagnostic delays and disease complications may occur as a consequence of low disease awareness and the lack of pathognomonic markers.
The pathogenesis of the multigenic autoinflammatory diseases are complex and, in some cases, precise pathogenesis is not clearly understood. Therefore, further research is warranted to understand the pathogenesis of these diseases.
Although conventional treatments are commonly used, biologics have shown promising results in the management of multigenic autoinflammatory diseases. Biologics targeting proinflammatory cytokines including IL-1, IL-6, TNF-α, IL-17A and IL-18 have been shown to ameliorate signs and symptoms of different multigenic autoinflammatory diseases.
Footnotes
Acknowledgements
Medical writing support was provided by Rajeeb Ghosh, PhD, Novartis Healthcare, and was funded by Novartis Pharmaceuticals Corporation. This manuscript was developed in accordance with Good Publication Practice (GPP3) guidelines. Authors had full control of the content and made the final decision on all aspects of this article.