
Abstract
Background:
Achondroplasia is the most common short-limbed skeletal dysplasia resulting from gain-of-function pathogenic variants in fibroblast growth factor receptor 3 (FGFR3) gene, a negative regulator of endochondral bone formation. Most treatment options are symptomatic, targeting medical complications. Infigratinib is an orally bioavailable, FGFR1–3 selective tyrosine kinase inhibitor being investigated as a direct therapeutic strategy to counteract FGFR3 overactivity in achondroplasia.
Objectives:
The main objective of PROPEL is to collect baseline data of children with achondroplasia being considered for future enrollment in interventional studies sponsored by QED Therapeutics. The objectives of PROPEL 2 are to obtain preliminary evidence of safety and efficacy of oral infigratinib in children with achondroplasia, to identify the infigratinib dose to be explored in future studies, and to characterize the pharmacokinetic (PK) profile of infigratinib and major metabolites.
Design:
PROPEL (NCT04035811) is a prospective, noninterventional clinical study designed to characterize the natural history and collect baseline data of children with achondroplasia over 6−24 months. PROPEL 2 (NCT04265651), a prospective, phase II, open-label study of infigratinib in children with achondroplasia, consists of a dose-escalation, dose-finding, and dose-expansion phase to confirm the selected dose, and a PK substudy.
Methods and analysis:
Children aged 3−11 years with achondroplasia who completed ⩾6 months in PROPEL are eligible for PROPEL 2. Primary endpoints include treatment-emergent adverse events and change from baseline in annualized height velocity. Four cohorts at ascending dose levels are planned for dose escalation. The selected dose will be confirmed in the dose-expansion phase.
Ethics:
PROPEL and PROPEL 2 are being conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, principles of the Declaration of Helsinki, and relevant human clinical research and data privacy regulations. Protocols have been approved by local health authorities, ethics committees, and institutions as applicable. Parents/legally authorized representatives are required to provide signed informed consent; signed informed assent by the child is also required, where applicable.
Discussion:
PROPEL and PROPEL 2 will provide preliminary evidence of the safety and efficacy of infigratinib as precision treatment of children with achondroplasia and will inform the design of future studies of FGFR-targeted agents in achondroplasia.
Registration:
ClinicalTrials.gov: NCT04035811; NCT04265651.
Introduction
Achondroplasia is the most common short-limbed skeletal dysplasia, affecting an estimated one in 15,000 to one in 30,000 live births worldwide, with an estimated global prevalence of 250,000.1,2 Characteristic clinical features of achondroplasia include disproportionate short stature (Table 1), smaller than average chest, macrocephaly with frontal bossing, midface hypoplasia, abnormal curvature of the spine, hypermobile joints, leg bowing, and shortening of the fingers and toes. 3 Individuals with achondroplasia experience a variety of physical, functional, and psychosocial complications and challenges.4,5 Common problems experienced during the first year of life include delayed motor development and hypotonia. 6 Recurrent otitis media and bowed legs are often seen in childhood as a result of abnormal skeletal growth. 7 Of greater concern are sleep apnea and sudden death that result from foramen magnum stenosis. 6 In later years, complications resulting from lumbosacral spinal stenosis can occur, including pain, muscle weakness, and paralysis. 7
Growth velocity in children with achondroplasia compared with those of average stature. 8 .

Achondroplasia is characterized by defective endochondral ossification resulting from gain-of-function pathogenic variants in the fibroblast growth factor receptor 3 (FGFR3) gene,9,10 which is a negative regulator of endochondral bone formation. FGFR3 is particularly prevalent on the surface of chondrocytes that give rise to cartilaginous bone. 11 Longitudinal bone growth is driven by the proliferation and differentiation of chondrocytes in the growth plate. Activating pathogenic variants of FGFR3 cause inhibition of chondrocyte proliferation and differentiation. 10 Achondroplasia results in most cases from either a G to A or G to C substitution at nucleotide position 1138 in FGFR3. 10 Both pathogenic variants result in the same glycine to arginine amino acid (Gly380Arg) substitution in the transmembrane domain of FGFR3; notably, 80% of affected individuals have a de novo event.
Management of the condition is mostly supportive in nature and may involve painful interventions aimed at preventing or treating complications of achondroplasia.5,12 Management varies from country to country, which prompted the recent development of an international consensus statement to standardize management and improve patient outcomes. 12
Infigratinib is an orally bioavailable and selective FGFR1–3 selective tyrosine kinase inhibitor in development for FGFR-related conditions, including cholangiocarcinoma and bladder cancer, in which FGFR2 and FGFR3 alterations have been implicated.13,14 Infigratinib inhibits phosphorylation of FGFR and, as a result, attenuates its downstream signaling, offering a direct therapeutic strategy to counteract the hyperactivity of FGFR3 in this condition. 5 Preclinical data in an Fgfr3Y367C/+ mouse model of achondroplasia showed that low doses of infigratinib (0.2, 0.5, and 2 mg/kg/day) reduced FGFR3 phosphorylation and restored activity of FGFR3 downstream signaling pathways to levels observed in wild-type mice.15,16 Infigratinib-treated mice exhibited substantial increases in the length of upper and lower limb long bones, and improvement in the shape and size of the foramen magnum, compared with untreated animals. 15
The PROPEL program was initiated to investigate the safety and efficacy of infigratinib in children with achondroplasia. Two studies are ongoing: The first is a study designed to observe children with achondroplasia over a minimum of 6 months (PROPEL study; NCT04035811), which is followed by the interventional PROPEL 2 study (NCT04265651) in which children who participated in PROPEL will receive treatment with infigratinib (Figure 1).

Design of the PROPEL and PROPEL 2 studies.
Methods
PROPEL
Study design
PROPEL is an ongoing, prospective, noninterventional clinical assessment study designed to collect serial assessments to characterize the natural history of achondroplasia symptoms in children for a minimum of 6 months and a maximum of 2 years (Figure 2). The study, which is sponsored by QED Therapeutics, Inc., is currently enrolling and will include up to 200 children. After the observational period, children may be eligible to enroll in the QED-sponsored interventional PROPEL 2 study.
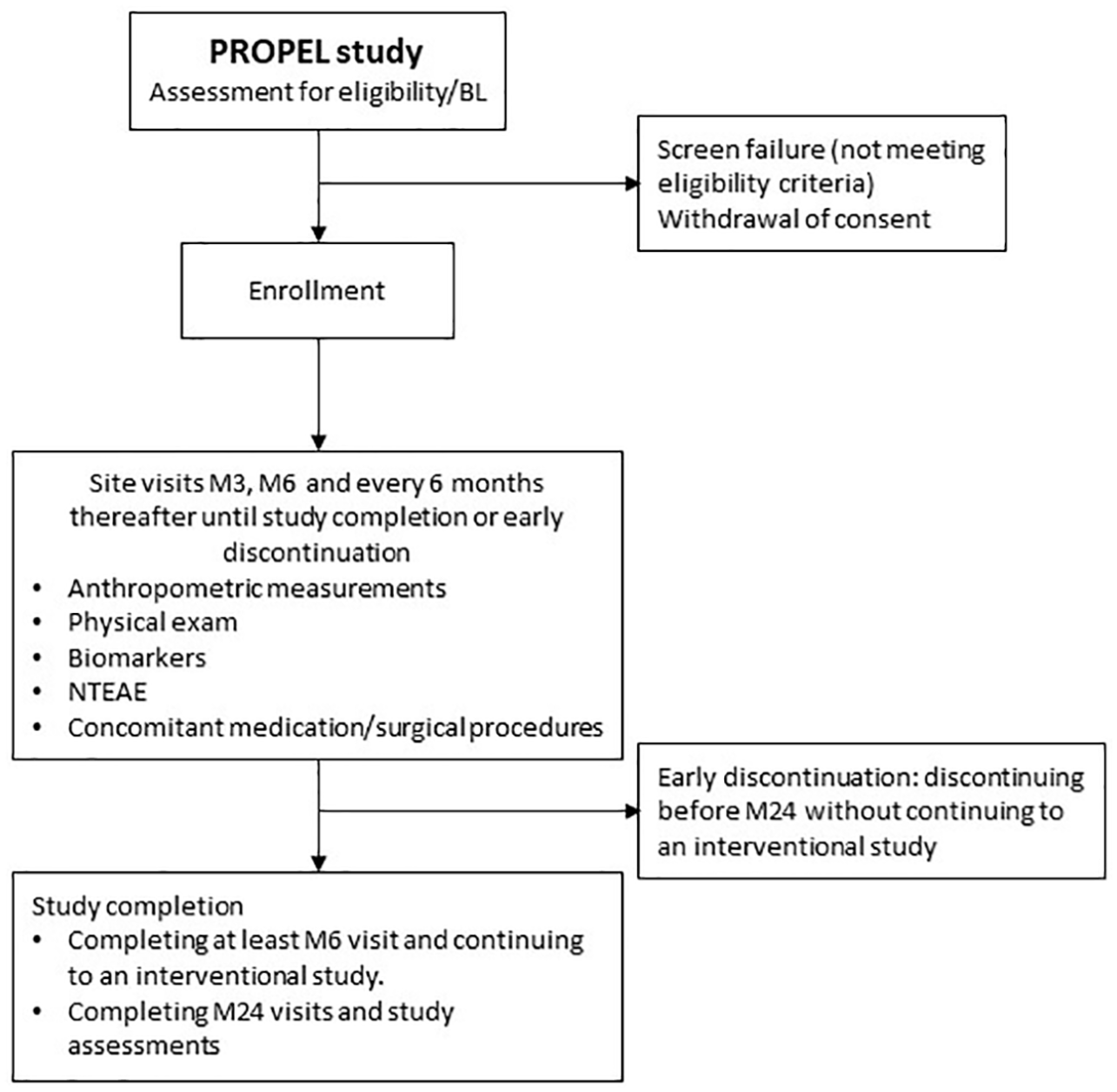
PROPEL study flowchart.
PROPEL is being conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, the principles of the Declaration of Helsinki, and all relevant human clinical research and data privacy regulations in the countries in which the study is being undertaken. The protocol has been approved by local ethics committees and institutional departments, as applicable.
Eligibility criteria
Eligibility criteria are summarized in Table 2. Eligible children are aged between 2.5 and 10 years (inclusive) at study entry, with diagnosis of achondroplasia. Parents or legally authorized representatives (LARs) are required to provide signed informed consent; signed informed assent by the child is also required, where applicable. Children must be ambulatory and able to stand without assistance. Participating children and their parents or LARs must be willing to comply with study visits and procedures.
PROPEL key inclusion and exclusion criteria.

ACH, achondroplasia; AHV, annualized height velocity; CNP, C-type natriuretic peptide; FGFR, fibroblast growth factor receptor; IGF, insulin-like growth factor 1; LAR, legally authorized representative; SD, standard deviation.
Exclusion criteria are hypochondroplasia or short stature condition other than achondroplasia, females who have had their menarche, height less than –2 or more than +2 standard deviations (SDs) for age and sex based on reference tables for growth in children with achondroplasia, and annualized height velocity (AHV) ⩽ 1.5 cm/year during the ⩾6 months before screening. Children are not allowed to participate if they have a concurrent disease or condition that, in the view of the Investigator and/or Study Sponsor, may affect growth or where the treatment is known to affect growth. Likewise, children with significant abnormality in screening laboratory test results will not be eligible to participate. Children will be excluded if they are receiving or have received treatment with growth hormone, insulin-like growth factor 1 (IGF-1), or anabolic steroids in the previous 6 months or long-term treatment (>3 months) at any time, as well as a C-type natriuretic peptide (CNP) analog, treatment targeting FGFR inhibition, or any other investigational product or investigational medical device for the treatment of achondroplasia or short stature at any time. Children who require regular long-term treatment (>1 month) with oral corticosteroids are not eligible, although low-dose ongoing inhaled steroid for asthma is acceptable. Children who have had previous limb-lengthening surgery are not eligible for inclusion.
Objectives and outcomes
The primary study objective is to collect baseline height velocity measurements of children with achondroplasia being considered for future enrollment in interventional studies sponsored by QED Therapeutics. The primary endpoint is to establish AHV.
Further objectives are collection of other baseline growth measurements of children with achondroplasia, exploratory evaluation of biomarker indicators of growth (e.g. type X collagen degradation fragment, collagen X marker), and assessment of achondroplasia-related medical events, such as obstructive sleep apnea, middle ear infections, and lumbar spinal stenosis, reported as medical history, or nontreatment adverse events (AEs). The study will also document achondroplasia-related surgical procedures (e.g. tympanostomy tube insertion and orthopedic procedures) that occurred before and during study participation.
Planned study period
The study was first posted on ClinicalTrials.gov on 29 July 2019 and the first child was enrolled in August 2019. It is active and enrolling participants as of 29 September 2021 (last update on ClinicalTrials.gov). The estimated study completion date is June 2026.
Statistical considerations
The sample size of approximately 200 children is considered enough to characterize the natural history of achondroplasia in children and will lead to sufficient enrollment in an interventional phase II or III study of infigratinib in children with achondroplasia. Relationships between selected baseline factors and height velocity may be assessed descriptively, and linear regression models may be used to assess the association between baseline factors and growth velocity. Descriptive statistics will be provided for demographics, subject disposition, and other assessments of bone and growth (biomarkers).
PROPEL 2
Study design
PROPEL 2 is a prospective, phase II, open-label study designed to provide preliminary evidence of safety and efficacy of oral infigratinib in children with achondroplasia, and to identify the dose of infigratinib to be explored in future studies (Figure 3). PROPEL 2 consists of a dose-escalation phase with extended treatment (n = 40), followed by a dose-expansion phase to confirm the selected dose and to provide evidence of efficacy (n = 20); a pharmacokinetic (PK) substudy is also included (approximately n = 18). In the dose-escalation portion of the study, children will be enrolled in ascending dose cohorts of approximately 10 children/cohort (four cohorts planned) and will be treated for 6 months at their assigned dose, continuing for an additional 12 months (extended treatment period) with dose modifications as required. Doses are weight based and will be adjusted for changes in weight approximately every 3 months. Cohort doses are as follows: cohort 1, 0.016 mg/kg per day; cohort 2, 0.032 mg/kg per day; cohort 3, 0.064 mg/kg per day; and cohort 4, 0.128 mg/kg per day. Children enrolled at the lowest dose levels (cohorts 1 and 2) may have their dose increased at month 6 and month 12 if there are no safety concerns and their AHV has not increased by at least 25% over baseline. The dose for further exploration will be selected based on safety and efficacy data from the four cohorts after all children have completed 6 months of treatment (dose-escalation phase). Once this dose has been selected, study participants will have their dose adjusted to the identified level. To support and confirm the dose identified for further study, approximately 20 new children will be enrolled into the dose-expansion phase and will receive 12 months’ infigratinib treatment at the identified dose. PROPEL 2 assessments are shown in Supplementary Table 1.

PROPEL 2 study flow chart.
The PK substudy will enroll approximately six children aged ⩾8 years in each of the dose cohorts (cohorts 2−4 only). Following the same visit schedule and assessments of safety and efficacy as those in the dose-escalation phase, children in each PK dose cohort will be treated and followed for 6 months at their assigned dose and, after the 6-month study visit, will continue treatment for an additional 12 months. Those in PK dose cohort 2 may have their dose increased to the next dose level at their 6- and 12-month study visits if no safety concerns have been identified and their AHV has not increased by at least 25% over baseline (a maximum of two dose increases is allowed). Children may also have their dose adjusted to the dose identified for further study at the time such a dose is determined, based on data from the dose-escalation phase. After completing the study, children will have the opportunity to continue treatment in an open-label extension study in which the safety and efficacy of long-term oral administration of infigratinib will be evaluated.
PROPEL 2 is being conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, the principles of the Declaration of Helsinki, and all relevant human clinical research and data privacy regulations in the countries in which the study is being conducted. The protocol has been approved by health authorities, local ethics committees, and institutional departments as applicable.
Eligibility criteria
Inclusion and exclusion criteria for PROPEL 2 are shown in Table 3. The same exclusion criteria for PROPEL are applicable in PROPEL 2. In addition, children with severe sleep apnea, children who have had guided growth surgery, or children who have had a recent fracture (within 6 months of screening) will also be excluded. Children receiving treatment with agents that are known to be strong inducers or inhibitors of cytochrome P450 3A4 and medications that increase serum phosphorus and/or calcium concentration will not be eligible to participate.
PROPEL 2 key inclusion and exclusion criteria.
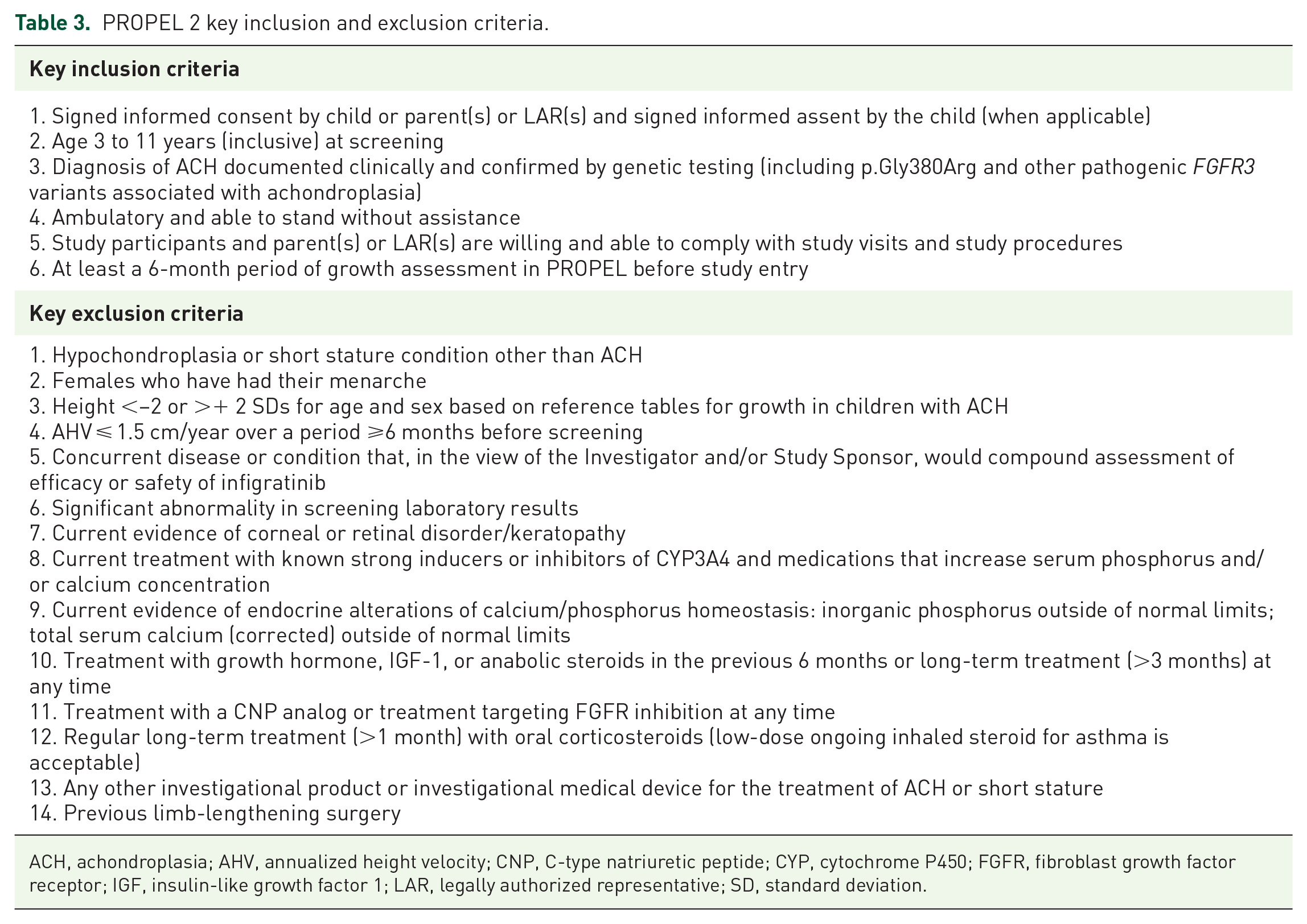
ACH, achondroplasia; AHV, annualized height velocity; CNP, C-type natriuretic peptide; CYP, cytochrome P450; FGFR, fibroblast growth factor receptor; IGF, insulin-like growth factor 1; LAR, legally authorized representative; SD, standard deviation.
Study oversight
A Data Review Committee (DRC) will monitor the safety and key efficacy data and provide recommendations to the Sponsor regarding dose escalation, dose de-escalation, and/or expansion of dose cohorts following criteria prespecified in the protocol based on the Bayesian optimal interval (BOIN) design 17 :
Cohort dose escalation: each cohort will commence after the safety of the prior dose cohort has been reviewed and confirmed by the DRC. During dose escalation, the opening of the next ascending dose cohort will be decided by the DRC based on review of safety data from approximately 10 children in each cohort after completion of ⩾4 weeks’ treatment and safety assessments.
Cohort dose de-escalation: the need for a cohort dose de-escalation will be determined by the DRC based on the safety assessment and incidence of treatment-emergent AEs (TEAEs) leading to dose reduction/discontinuation in an individual child. If at any point in the study, ⩾30% of children enrolled in a cohort meet the dose discontinuation or reduction criteria, then enrollment in that and/or any higher dose cohort (if applicable) will be paused, and the DRC will be convened. The DRC will determine if the dose of the current or higher cohort should be de-escalated.
Dose reduction/discontinuation for an individual participant: although the DRC will monitor safety and will consider the number of children meeting the dose-reduction/discontinuation criteria to determine whether a cohort dose escalation can proceed or whether a dose de-escalation is needed at the cohort level, dose modifications in an individual participant will be managed by investigators following criteria prespecified in the protocol.
Objectives and outcomes
The study objectives and endpoints are summarized in Table 4. The primary objective of the dose-escalation phase is to identify a dose of infigratinib for children with achondroplasia to be used for further study. The primary objective of the dose-expansion phase is to provide preliminary evidence of efficacy of infigratinib for the treatment of achondroplasia. The primary objective of the PK substudy will be to evaluate the PK profile of infigratinib and its major active metabolites after oral administration of infigratinib. Primary endpoints will be incidence of TEAEs that lead to dose reduction or drug discontinuation and change from baseline in height velocity (annualized to cm/year) in the dose-escalation phase and change from baseline in height velocity (annualized to cm/year) in the dose-expansion phase. The primary endpoints of the PK substudy will be PK parameters including peak plasma concentration (Cmax), last measurable plasma concentration (Clast), time to reach Cmax (Tmax), area under the plasma concentration–time curve over 24 h (AUC24), elimination half-life (T1/2), area under the plasma concentration–time curve from time zero to infinity (AUCinf), apparent total clearance of drug from plasma after oral administration (CL/F), apparent volume of distribution during terminal elimination phase after oral administration (Vz/F), and accumulation ratio (Racc). Baseline is defined as the AHV obtained after at least 6 months’ observation in the PROPEL study. Secondary objectives will be to evaluate the safety and tolerability of infigratinib in children with achondroplasia, to evaluate changes from baseline in anthropometric parameters (including sitting height and body proportions) after administration of infigratinib, and to evaluate the PK and pharmacodynamic profile of infigratinib in children with achondroplasia. Evaluation of changes in the burden of achondroplasia, including quality of life, is an exploratory endpoint.
PROPEL 2 objectives and endpoints.
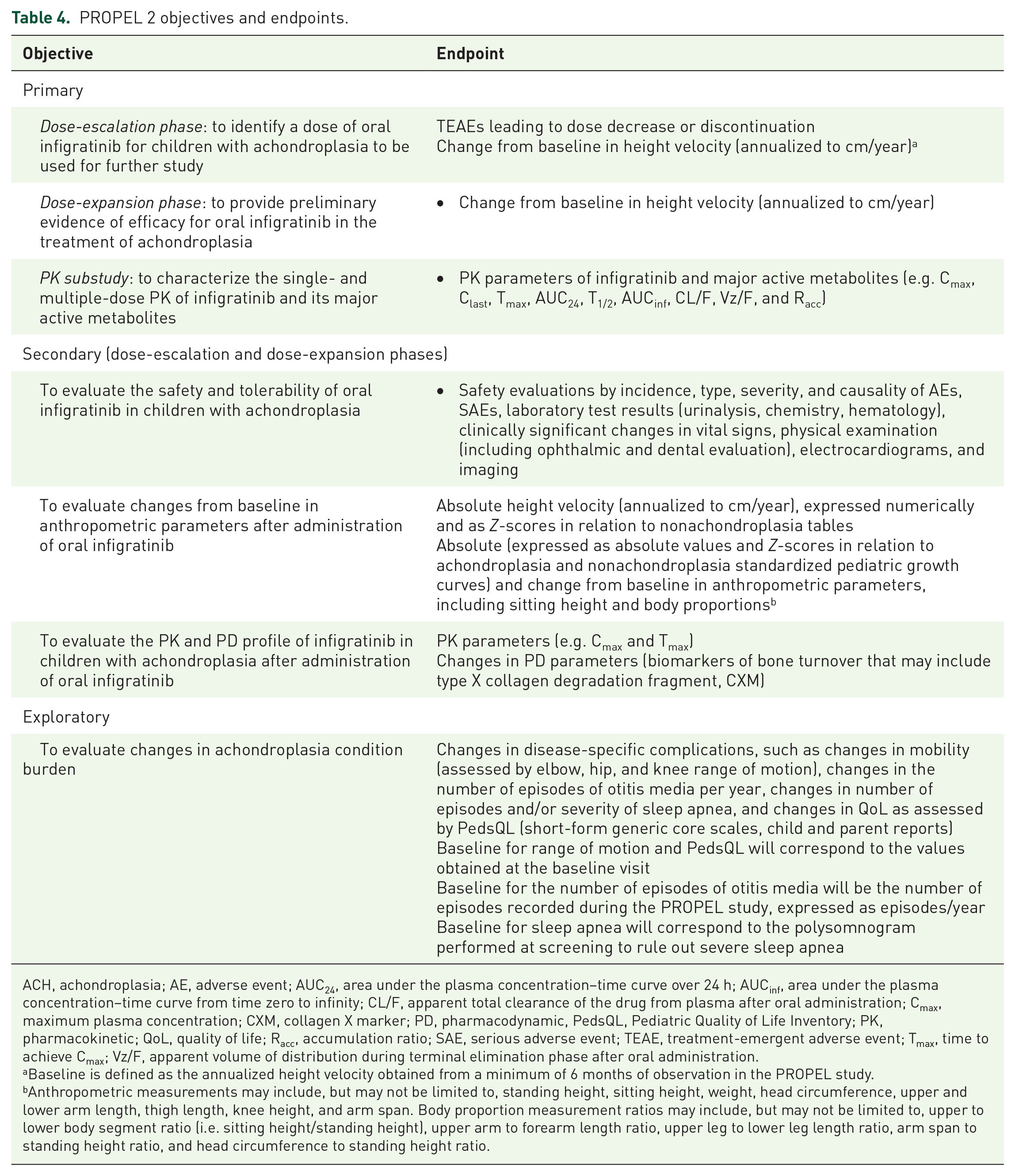
ACH, achondroplasia; AE, adverse event; AUC24, area under the plasma concentration–time curve over 24 h; AUCinf, area under the plasma concentration–time curve from time zero to infinity; CL/F, apparent total clearance of the drug from plasma after oral administration; Cmax, maximum plasma concentration; CXM, collagen X marker; PD, pharmacodynamic, PedsQL, Pediatric Quality of Life Inventory; PK, pharmacokinetic; QoL, quality of life; Racc, accumulation ratio; SAE, serious adverse event; TEAE, treatment-emergent adverse event; Tmax, time to achieve Cmax; Vz/F, apparent volume of distribution during terminal elimination phase after oral administration.
Baseline is defined as the annualized height velocity obtained from a minimum of 6 months of observation in the PROPEL study.
Anthropometric measurements may include, but may not be limited to, standing height, sitting height, weight, head circumference, upper and lower arm length, thigh length, knee height, and arm span. Body proportion measurement ratios may include, but may not be limited to, upper to lower body segment ratio (i.e. sitting height/standing height), upper arm to forearm length ratio, upper leg to lower leg length ratio, arm span to standing height ratio, and head circumference to standing height ratio.
Planned study period
The study was first posted on ClinicalTrials.gov on 11 February 2020 and the first dose was administered on 15 July 2020. PROPEL 2 is active and enrolling participants as of 3 August 2021 (last update on ClinicalTrials.gov). The estimated study completion date is January 2024.
Statistical considerations
Data will be summarized separately for children in the dose-escalation, dose-expansion, and PK substudy phases. The mean, SD, median, quartile 1 (Q1), quartile 3 (Q3), minimum, and maximum may be provided for continuous data at each timepoint. The number/count and percentage will be provided for categorical data.
Dose-escalation and de-escalation rules are based on the BOIN design 17 with a target toxicity rate of 25%. Selection of the dose for the expansion phase will be based on assessment of approximately 10 children per cohort. If a true AE incidence is 25%, 10 children per cohort will allow observation of at least of one AE with 94.4% confidence. With 10 children per cohort, there is also a 62.5% chance of obtaining a 95% confidence interval with a half-width that is at most 1.5 cm/year, assuming the change from baseline in height velocity follows a normal distribution and the SD is 2 cm/year.
In the dose-expansion phase, approximately 20 children will be enrolled at the selected dose level. An AHV increase of ⩽0.5 cm/year will be considered not clinically relevant and will be used as the null hypothesis. Assuming an increase in height velocity of 2 cm/year after initiation of infigratinib treatment, with an SD of 2 cm/year, 20 children will provide approximately 88.9% power to demonstrate that treatment with infigratinib can result in an increase from baseline in height velocity of >0.5 cm/year at a one-sided significance level of 0.025.
For the dose-escalation phase, all analyses will be performed separately for each dosing cohort based on the dose originally received and in total, taking into consideration dose increases and reductions. Children enrolled in the dose-expansion phase will be analyzed for both safety and efficacy.
A primary analysis for the dose-escalation phase is planned after all children have had the opportunity to complete 6 months of treatment. The primary analysis for the dose-expansion phase is planned after all children in this phase have had the opportunity to complete 12 months of treatment. In addition, other ad hoc analyses may be performed during the course of the study, as needed.
All safety analyses will be performed using the safety analysis set, defined as children who have received ⩾1 dose of study drug. Analyses of growth parameter endpoints will be performed for children with a baseline and ⩾1 postbaseline growth parameter assessment.
Discussion
The impaired endochondral ossification characteristic of achondroplasia results from a dominantly inherited FGFR3 pathogenic variant. Following on from the development of selective FGFR inhibitors with demonstrated efficacy in the treatment of patients with FGFR-related conditions, targeting FGFR3 mutations in achondroplasia represents a promising strategy for this challenging condition. Preclinical data provided early and encouraging data for infigratinib, an FGFR1–3 inhibitor, where treatment with infigratinib for 15 days (day 1–16) revealed dose-dependent improvements in foramen magnum size and shape and long bone length in Fgfr3Y367C/+ mice. 16 The PROPEL study program was therefore initiated to study infigratinib, an investigational, orally available, small molecule targeting FGFR as a novel approach to the treatment of children with achondroplasia.
The PROPEL study aims to characterize the natural history of achondroplasia in a cohort of affected children who can subsequently participate in an interventional study with oral infigratinib. It is designed to establish baseline data in participants, including growth velocity for children with achondroplasia before the start of treatment. Establishing the natural history of the condition is essential in rare diseases to avoid uncertainty regarding the optimal timing of intervention, incorrectly attributing side effects to the study drug rather than as disease-related complications (or vice versa), and misunderstanding which are the best outcome measures. 18 Specifically, by establishing AHV in PROPEL, each child’s change in AHV in PROPEL 2 following infigratinib treatment can be compared with PROPEL data, allowing each child to act as their own control. Therefore, natural history observational studies, such as PROPEL, facilitate the assessment of outcomes of greatest relevance to individuals with a specific condition and rigorous assessment of safety data. 18
PROPEL 2 is the first study to target the genetic driver of overactivity in FGFR3 and its downstream signaling pathways in children with achondroplasia. Infigratinib has proven efficacy and tolerability in the treatment of cholangiocarcinoma and bladder cancer, other conditions characterized by FGFR alterations.14,19 Notably, doses used for oncological indications are substantially higher than the doses used in the PROPEL 2 study. The properties of low doses of infigratinib in the pediatric population with achondroplasia and an appropriate therapeutic dose remain to be elucidated. The safety monitoring of this study takes into account the known safety profile of higher doses of infigratinib in patients with cholangiocarcinoma and bladder cancer, in which settings hyperphosphatemia was the most common known on-target toxicities.14,19 In the achondroplasia setting, however, infigratinib doses are considerably (10–100 times) lower than those used in the cholangiocarcinoma and bladder cancer settings, with the result that these toxicities are not expected to occur in PROPEL 2. Preclinical experiments conducted to date support this hypothesis and showed that hyperphosphatemia does not occur at the low doses of infigratinib that show activity in vivo.15,20
The outcomes of PROPEL 2 were chosen to reflect not only increase in height but also a range of wider health measures considered to be of importance, including quality of life and medical complications. Quality of life – in particular functional domains – can be compromised in children with achondroplasia21,22 and a focus on aspects other than height is likely to provide a rounded assessment of treatment success in individuals affected by achondroplasia.
To date, only one drug targeting the underlying causes of achondroplasia has been approved, with additional agents currently in development for this indication. Vosoritide is a CNP analog, administered by daily subcutaneous injection, which inhibits the FGFR3-mediated MAPK signaling pathway,23,24 and was approved in the European Union in August 2021 for children with achondroplasia from age 2 years until growth plate closure. In the United States, vosoritide for injection has been granted accelerated approval by the US Food and Drug Administration to increase linear growth in pediatric patients with achondroplasia who are aged 5 years and older with open epiphyses (growth plates). The approval of vosoritide for the treatment of children with achondroplasia is paving the way for new options in the management of this condition. To the best of our knowledge, infigratinib is the only agent currently in clinical development for the treatment of children with achondroplasia that is orally administered.
Conclusions
Children with achondroplasia currently have few treatment options; however, the development of agents that effectively target the underlying cause of the condition, namely, FGFR3 mutations, has the potential to improve outcomes in this setting. The PROPEL study described in this article is an ongoing observational study investigating the natural history of children with achondroplasia, providing a basis against which the success of any subsequent intervention can be measured. The follow-up phase II PROPEL 2 study is designed as a dose-finding study and will obtain preliminary evidence of safety and efficacy. It will assess the effects of infigratinib on growth, development, and disease burden in children with achondroplasia, as well as its safety in this population. It is the first study to use a small molecule, orally administered FGFR inhibitor in this setting. PROPEL and PROPEL 2 are expected to inform the design of future studies of this promising new targeted approach to the treatment of children with achondroplasia. Successful development and approval of agents with differing modes of action could provide children with achondroplasia with a range of potentially life-altering treatment options.
Supplemental Material
sj-docx-1-tab-10.1177_1759720X221084848 – Supplemental material for Infigratinib in children with achondroplasia: the PROPEL and PROPEL 2 studies
Supplemental material, sj-docx-1-tab-10.1177_1759720X221084848 for Infigratinib in children with achondroplasia: the PROPEL and PROPEL 2 studies by Ravi Savarirayan, Josep Maria De Bergua, Paul Arundel, Helen McDevitt, Valerie Cormier-Daire, Vrinda Saraff, Mars Skae, Borja Delgado, Antonio Leiva-Gea, Fernando Santos-Simarro, Jean Pierre Salles, Marc Nicolino, Massimiliano Rossi, Peter Kannu, Michael B. Bober, John Phillips, Howard Saal, Paul Harmatz, Christine Burren, Garrett Gotway, Terry Cho, Elena Muslimova, Richard Weng, Daniela Rogoff, Julie Hoover-Fong and Melita Irving in Therapeutic Advances in Musculoskeletal Disease
Footnotes
Acknowledgements
The authors would like to thank all the children participating in these studies, their families, the clinical investigators, subinvestigators, study coordinators, clinical research nurses, and other team members at the investigational sites, clinical research monitors, data management team, and all other members of the PROPEL and PROPEL 2 study teams. We would also like to thank Lee Miller and Deirdre Carman at Miller Medical Communications for their medical editing and writing assistance, which was funded by QED Therapeutics.
Author contributions
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RS: reports grant support from Ascendis, BioMarin, QED and Theracon, received consulting fees from BioMarin and was a paid advisory board member for Ascendis, BioMarin, QED and Sanofi. JMDB: has nothing to disclose. PA: has nothing to disclose. HM: has nothing to declare. VC-D: was a paid advisory board member for BioMarin. VS: has nothing to disclose. MS: has nothing to disclose. BD: has nothing to disclose. AL-G: has nothing to disclose. FS-S: was a paid advisory board member for BioMarin. JPS: has nothing to disclose. MN: has nothing to disclose. MR: was a paid advisory board member for BioMarin. PK: has nothing to disclose. MB: reports grant support and paid consultancies from Ascendis Pharma, BioMarin, Pfizer, QED Therapeutics. JPIII: has nothing to disclose. HS: reports grant support from Ascendis Pharma, BioMarin, Pfizer, QED Therapeutics. PH: reports paid consutancies for Audentis, Aeglea, Homology, BioMarin, Shire, Genzyme, Ultragenyx, JCR, Denali, Orphazyme, Inventiva, Paradigm, REGENXBIO, Sangamo, QED, Ascendis Pharma, contracts for research from BioMarin and Inventiva and honoraria from BioMarin, Shire, Genzyme, Ultragenyx, and Ophazyme. CB: reports research support from Amgen, Pfizer, QED Therapeutics. GG: has nothing to disclose. TC, EM, RW, DR: report that they are employees of QED therapeutics. JH-F: reports paid consultancies from Pfizer/Therachon, BioMarin, QED Therapeutics, Sanofi, Ascendis Pharma, and grant support fromPfizer/Therachon, BioMarin, Ascendis Pharma. MI: was a paid advisory board member for Ascendis Pharma, BioMarin, QED Therapeutics, Sanofi, Therachon/Pfizer.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by QED Therapeutics, an affiliate of BridgeBio.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.