
Abstract
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) is an autosomal recessive disorder characterized by hypophosphatemia, rickets, hyperphosphaturia, elevated 1,25(OH)2D, and hypercalciuria. Mutations in SLC34A3, the gene encoding the sodium-dependent cotransporter NPT2c, have previously been described as a cause of HHRH. Here, we describe two male siblings with rickets and hypercalciuric nephrolithiasis born to unrelated parents, and their response to oral phosphate supplementation and growth hormone therapy. Whole exome sequencing of the oldest brother, and polymerase chain reaction and Sanger sequence analysis of the identified SLC34A3 mutations, was performed for confirmation and to evaluate his siblings and parents. Serum and urine biochemical parameters of mineral homeostasis before and after therapy were evaluated. Whole exome sequencing analysis identified a previously reported heterozygous deletion SLC34A3.g.2259-2359del101bp on the maternal allele, and a novel heterozygous single nucleotide deletion SLC34A3.c.671delT on the paternal allele of the two affected brothers. The parents and the unaffected brother are heterozygous carriers. Recombinant human growth hormone (rHGH) plus oral phosphate in one affected brother improved the renal phosphate leak and resulted in accelerated linear growth superior to that seen with oral phosphate supplementation alone in the other affected brother. Our case study is the first to demonstrate that rHGH can be considered in addition to oral supplementation with phosphorus to improve linear growth in patients with this disorder, and suggests that renal phosphate reabsorption in response to rHGH is NPT2c-independent.
Introduction
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH, OMIM 241530) is a rare autosomal recessive disease, first described by Tieder in 1985. 1 In HHRH patients, fibroblast growth factor 23 (FGF23) levels are suppressed, which sets this disorder apart from FGF23-dependent hypophosphatemias such as X-linked hypophosphatemic rickets (XLH, OMIM 307800), 2 and autosomal dominant hypophosphatemic rickets (ADHR, OMIM 193100). 3 As a result, markedly increased circulating 1,25-dihydroxy-vitamin D [1,25(OH)2D] stimulates absorption of calcium from the gut, which suppresses parathyroid hormone (PTH) and causes hypercalciuria. 4
Mutations in SLC34A3, the gene encoding the sodium-dependent cotransporters NPT2c, cause classical HHRH, whereas several phenotypically similar syndromes were recently attributed to mutations in the related transporter SLC34A1/NPTa.2,5–7 Carriers of one mutant SLC34A3/NPT2c transporter allele may develop idiopathic hypercalciuria along with mild hypophosphatemia, but bone disease is generally absent.7,8 Conventional therapy of HHRH consists of oral phosphate supplementation. Active vitamin D analogues should be avoided to not further worsen the hypercalciuria.
Growth hormone (GH) and insulin-like growth factor 1 (IGF-1) play an important role in Ca and phosphate homeostasis during childhood and adolescent growth to meet the demands of accelerated bone formation. Recombinant human growth hormone (rHGH) therapy has been used with some benefit in patients with XLH and the Hyp mouse model of XLH.9,10 This may be due, in part, to the calcium- and phosphate-sparing effects of GH and IGF1 in the kidneys.11–13 In seven children with idiopathic GH deficiency, intestinal calcium absorption increased during rHGH administration, resulting in a compensatory decrease of PTH and serum 1,25(OH)2D, and, thus, was interpreted as direct effect of rHGH at the intestinal lining. 14 IGF1 increases expression of sodium-dependent phosphate transport in the renal brush border of rabbits and hypophysectomized rats,15,16 and stimulates renal 1-alpha-hydroxylation of 25-hydroxy-vitamin D [25(OH)D] independently from PTH in parathyroidectomized dogs. 12 IGF1 directly increased levels of NPT2a in the plasma membrane of opossum kidney and of human cultured proximal tubule cells.17,18 It is unknown whether GH/IGF1 regulates NPT2c. GH/IGF-1-induced calcitriol production increases intestinal calcium absorption via the intestinal epithelial calcium channel TRPV6. In the kidney, calcitriol controls Ca reabsorption through TRPV5 expression in distal tubule cells. 19 This results in a positive calcium balance necessary for accelerated growth.
Case report
The index case (II-1) is a boy aged 9 years and 10 months, who initially presented at age 2 years with poor growth, bowing of legs, bone pain, and radiological changes consistent with rickets and a kidney stone, but no phosphate supplements were given, and he was lost to follow up. At this presentation, his height was at the 3rd percentile, and weight at the 10th percentile. He had Tanner I puberty. His right tibia displayed internal torsion of about 30°, and genu varum. Laboratory testing revealed normal serum calcium, but low serum phosphate, and elevated alkaline phosphatase (Figure 1). His 25(OH)D was low, but 1,25(OH)2D was elevated, whereas intact PTH and c-terminal FGF23 were appropriately reduced. Urine tubular reabsorption of phosphate (%TRP) was reduced, and the urine calcium/urine creatinine ratio was elevated. Oral phosphate supplementation with sodium/potassium phosphate (Neutra-Phos) 1000 mg/day was initiated, but rickets progressed, despite reported good compliance. He started puberty at 10 years 6 months, but did not experience a growth spurt. He required corrective orthopedic surgeries at 11 years 11 months, and at 13 years 6 months (Figure 2). By 16 years, he had reached his final adult height of 148.1 cm (0.08 percentile, or −3.14 SD; Figure 3). His upper/lower segment ratio was 1.14. rHGH therapy was considered, but not started. At 12 years, II-1 presented with flank pain and left hydronephrosis due to an obstructing left ureter stone, requiring lithotripsy. He subsequently developed further bilateral asymptomatic kidney stones.

Pedigree with genotypes and initial biochemical and clinical findings in the family.
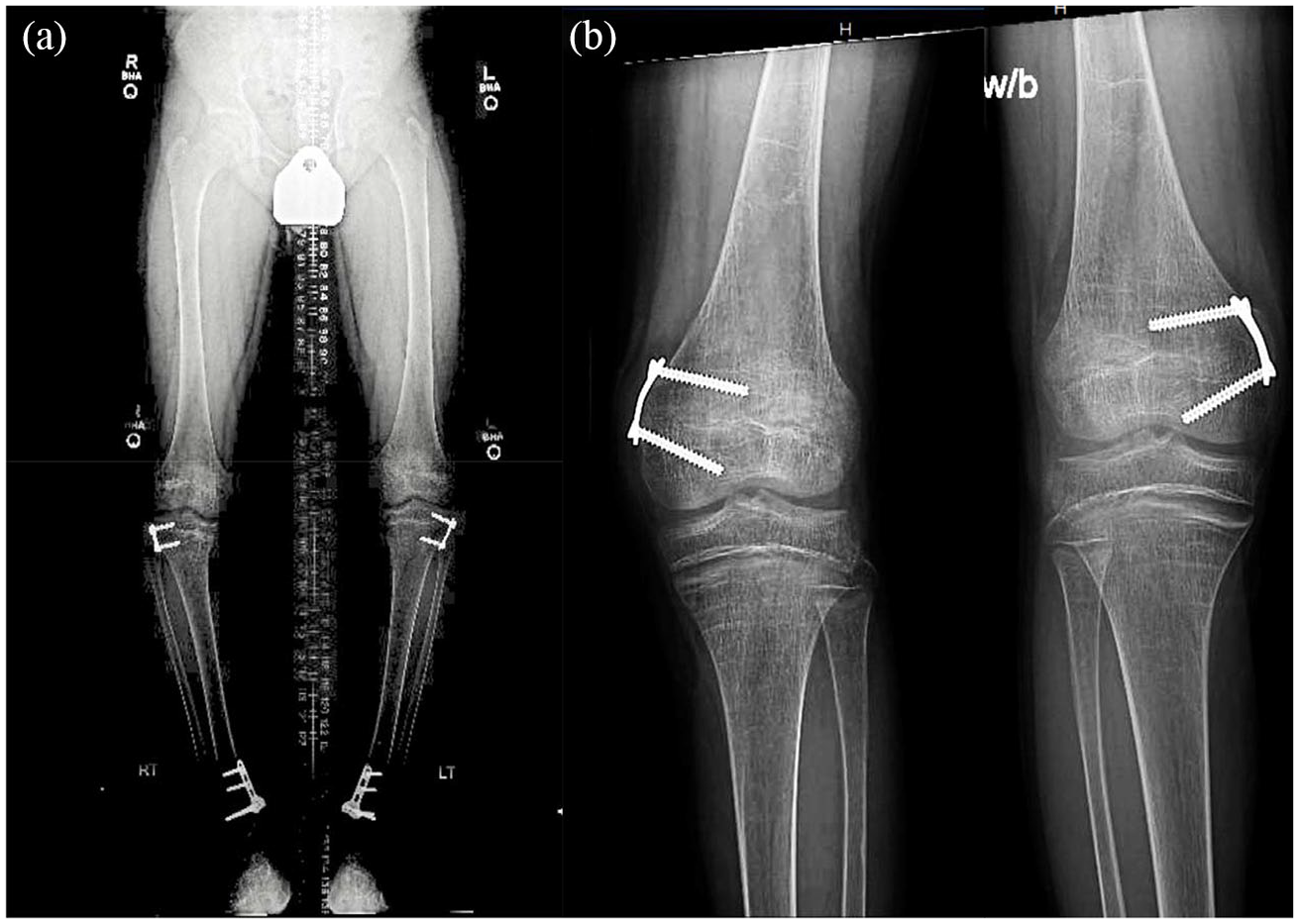
(a) Radiological bilateral full leg images of II-1 (Index) at age 12 years 3 months demonstrate the extent of corrective surgeries and the hardware left in place. (b) Radiological images of both knees of II-2 at age 13 years 6 months after bilateral distal medial hemiepiphysiodesis; notice diffuse osteopenia and metaphyseal irregularity.

(a) Height chart of II-1 (Index) shows short stature and poor growth velocity. 1, epiphysiodesis and placement of supramalleolar pin, removed after 6 weeks; 2, epiphysiodesis and placement of permanent hardware in bilateral distal, and proximal tibia. (b) Height chart of II-2 shows short stature and poor linear growth with no significant improvement with phosphate treatment, and acceleration after the rHGH initiation (star). His bone age (triangles) was delayed compared to chronological age. 1, hemiepiphysiodesis and permanent hardware placed in bilateral distal femur. (c) Height chart of II-3, who is heterozygous.
The middle brother (II-2) at age 8 years was also found to have hypophosphatemia along with elevated alkaline phosphatase, hypercalciuria, low serum PTH, and elevated 1,25(OH)2D (Figure 1) and was started on oral phosphate supplementation with sodium/potassium phosphate (neutral-Phos) 1000 mg/day 4 months later. Notwithstanding, he developed knee pain and bowing of both legs, and, at the age of 13 years 6 months, he underwent bilateral distal femoral medial hemiepiphysiodesis (Figure 2). At 12 years and 6 months, his height remained at the 0.3rd percentile with poor growth (3.4 cm/year) despite normal start of puberty (Tanner stage II). He had normal thyroid function and serum IGF1 levels. His bone age was 11 years (Figure 3). At age 12 years and 4 months rHGH 0.3 mg/kg/week was started, and growth velocity improved to 8.5 cm/year, resulting in a height improvement from 0.3rd to 1.52nd percentile after 15 months. On rHGH his %TRP improved from 83.5% to 96.5% (Table 1). At age 14 years, II-2 developed nonobstructive right renal nephrolithiasis, and passed a stone.
Comparison of biochemical findings in II-2 on treatment with phosphate only, and 15 months after rHGH was added.

FGF23, fibroblast growth factor 23; IGF-1, insulin-like growth factor 1; rHGH, recombinant human growth hormone; RU, relative units; TRP, tubular reabsorption of phosphate.
His youngest sibling (II-3) remained unaffected and grew well along the 90th percentile, and at age 10 years his height was at +1.7 SD. The father (I-1) passed a kidney stone at age 41 years. His height is 155 cm (< 1st percentile, −2.8 SD), and he has no history of rickets, and no obvious skeletal deformities. The mother (I-2) passed a kidney stone at age 43 years. Her height is 162.5 cm (50th percentile, 0 SD), and she likewise lacks history of rickets or skeletal deformities. The parents are not related.
Results
Genetic evaluation was performed following informed written consent of all family members (Yale HIC1501015216) using whole exome sequencing of the index case II-1 revealed compound heterozygosity for a previously reported mutation: SLC34A3.g.2250-2359del101bp and one novel mutation: SLC34A3.c.671delT. 4 No disease-causing variants were detected in SLC34A1, PHEX, DMP1, ENPP1, or FGF23. Additionally, the previously reported heterozygous variants SNPs het.SLC34A3.c.T757C:p.L253L [minor allele frequency (MAF): 0.6281] and het.SLC34A3.c.A1538T:p.E513V (MAF: 0.9994) and a homozygous variant hom.SLC34A1c.T774C:p.H258H (MAF: 0.495) were observed.4–7 Polymerase chain reaction-based Sanger sequencing analysis of family members revealed that SLC34A3.c.671delT was inherited from the father (I-1) and SLC34A3.g.2250-2359del101bp was inherited from the mother (I-2). The affected brother (II-2) likewise carried both mutations, whereas the unaffected brother carried only SLC34A3.g.2259-2359del101bp from the mother (Figure 4). SLC34A3.c.671delT has not been reported previously in dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), the Exome variant server (http://evs.gs.washington.edu/EVS), or the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/all.php). Deletion of c.671delT is predicted to result in a frameshift with loss-of-function of the NPT2c sodium-phosphate cotransporter.

(a) Sanger sequencing results of SLC34A3 g.2259-2359 del101. Black rectangle contains 101 base pair (bp) deletion (Geneious 6.0.6 https://geneious.com). Green rectangle contains the duplication that appears in the flanking region. (b) Polymerase chain reaction products separated by 2% agarose Tris-acetate-EDTA gel electrophoresis to visualize the 101 bp deletion in I-2, II-1, II-2, and II-3 in comparison with I-1, who does not have this deletion (c) Sanger sequencing results of SLC34A3 c.671delT. Rectangle contains 1 bp deletion.
Discussion
Here, we report two siblings with HHRH who are compound heterozygous for a novel SLC34A3.c.671delT mutation and the previously reported deletion SLC34A3.g.2259-2359del101bp in several cases with HHRH.4,6,7 Segregation analysis was consistent with the autosomal recessive inheritance of HHRH. Furthermore, in silico analysis predicts a deleterious phenotype of these compound heterozygous mutations, which explains the renal phosphate wasting syndrome of the two brothers. Oral phosphate supplements, which are considered standard therapy in HHRH, improved their elevated 1,25(OH)2D levels and the hypercalciuria, but this was not enough to cure rickets, short stature, and to reduce the risk for nephrocalcinosis.
Growth retardation is more common in patients with FGF23-dependent hypophosphatemias and refractory to therapy with phosphate and calcitriol. Pretreatment height Z scores of −6.5 to −1.0 were reported in 21 Norwegian subjects with XLH, and remained essentially unchanged with −6.3 to −0.8 after 1 year of standard therapy. 20 Likewise, individuals with the less common ADHR, and autosomal recessive hypophosphatemic rickets can present with growth retardation.5,21 This is thought to be due to FGF23 directly reducing proliferation of growth plate chondrocytes. 22 Unlike the FGF23-dependent forms, most individuals with HHRH reach normal adult height with improved metabolic control alone. 7 Growth hormone therapy can be beneficial in children with XLH, 9,23,24 and in the Hyp mouse model of XLH. 10 Haffner and colleagues found improved growth rate and median adult height in three children in whom rHGH was initiated before puberty for 3.1−6.3 years. However, the upper/lower ratio was exaggerated in some. 22 In a randomized controlled open–label study of 16 children with XLH, height increased by 0.7 SDS when compared with untreated individuals, but body proportion was not affected. 24
Bone pain and hypercalciuria of both affected brothers improved on phosphate supplementation, but their rickets did not heal completely, and their growth rate did not improve significantly. Although their predicted target height was projected at the 22nd percentile, our index case and his brother II–2 remained at the 3rd and 0.5th percentile, respectively. Their weight curves were different: the index case gained weight rapidly between ages 10 and 14, with his body mass index reaching 90th percentile, and his skeletal maturation was accelerated. His brother II-2 had less weight gain, possibly due to anorexia induced by psychostimulant therapy for attention deficit disorder. Addition of rHGH to standard therapy of brother II-2 resulted in acceleration of growth. Interestingly, his renal handling of phosphate also normalized compared with that of II-1, who remained on standard therapy alone. Normalization of TRP is consistent with the previously reported GH or IGF-1 action at the proximal tubules and may have contributed to the treatment success of rHGH in II-2 in addition to its direct action on growth plates. Efficacy of rHGH in the presence of a NPT2c loss-of-function mutations furthermore suggests that GH or IGF-1 may act through upregulation of NPT2a (or another Pi transporter different from NPT2c) at the proximal tubules.
Intestinal calcium absorption and renal calcium excretion in children depends largely on skeletal demands during periods of rapid growth. Urinary calcium is low in infants, and increases gradually during childhood, reaching adult levels by the end of pubertal growth. 25 In situations of GH hypersecretion, and rHGH therapy of non-GH deficient children, renal calcium excretion in human subjects is modified. However, the renal action of GH/IGF-1 is difficult to evaluate because of parallel changes in intestinal calcium absorption, and, consequently, in filtered calcium loads. In a healthy individual during pubertal growth spurt, rapidly rising IGF-I upregulates calcitriol via stimulation of 1-α hydroxylase in the proximal tubule. While excess IGF-1 causes increased levels of 1,25(OH)2D and increased intestinal absorption of calcium in intestine through the epithelial calcium channel TRPV6, renal calcium reabsorption is stimulated through up-regulation of the epithelial calcium channel TRPV5 in the distal tubule, and prevents calcium wasting. 19 This may explain why we did not see increased urinary calcium in our rHGH-treated subject. Whether this is true in all adolescents with HHRH is not known.
Although this report is limited by its small sample size, the two genetically identical brothers offered a unique opportunity to compare response to standard therapy with or without rHGH therapy. Surgical intervention and inconsistent compliance with phosphate supplementation in the older brother II-1 could have contributed to his growth failure. Conversely, start of II-2 on phosphate supplementation 18 months earlier than the index case, and his 12 months delay in onset of puberty, may have resulted in improved response to standard therapy regardless of rHGH. At the time of this report, II-2 was still growing, and it remains to be seen if GH therapy improves his final adult height. HHRH is a very rare condition (prevalence 1:250,000), which unfortunately makes it unlikely that a prospective, randomized study of rHGH therapy for growth failure in these patients will ever be possible. This case report, however, should raise awareness, so that pediatric endocrinologists treating children with HHRH may consider this treatment option. In conclusion, we report a novel SLC34A3.c.671delT mutation in two siblings, affected by HHRH, and their father. The two siblings, who were compound heterozygous, had rickets and nephrolithiasis. The parents, with heterozygous but with different mutations in SLC34A3, had nephrolithiasis but no rickets. So far, II-3 has had no signs of rickets or nephrolithiasis. To our knowledge, brother II-2 is the first subject with HHRH to be treated with phosphate and rHGH, which significantly improved his growth velocity. Improved renal handling of phosphate suggests that this action of rHGH and IGF-1 is independent of NPT2c.
Footnotes
Acknowledgements
We are grateful to the Yale O’Brien Center (Pilot grant to C.B., NIH P30DK079310).
Contributor’s Statement
D. D. identified, evaluated, and treated study subjects, obtained DNA, drafted the initial manuscript. C. B. and A. C. performed DNA analysis, provided the molecular diagnosis, and revised and critically edited the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The authors declare that there is no conflict of interest.