
Abstract
Hypophosphatasia is a rare inherited disease caused by a loss of function mutations in the gene that codes for the tissue-nonspecific alkaline phosphatase enzyme. It is autosomally inherited and at least 388 different genetic defects have been identified. The clinical presentation is variable from a severe perinatal form, that is fatal if untreated, to adult-onset disease. This review covers the pathophysiology, diagnosis and current management option including the recently licensed enzyme replacement therapy asfotase alfa.
Introduction
Hypophosphatasia (HPP) is a rare inherited disease caused by a loss of function of the enzyme tissue-nonspecific alkaline phosphatase (TNSAP). There are a number of challenges for the clinician in treating patients with this condition. Firstly, there is great heterogeneity in the clinical presentation and severity of the disease among affected individuals. A milder form of the disease that presents in adulthood can be misdiagnosed as osteoporosis if the treating doctor is not familiar with this condition. 1 Moreover, the imperfect correlation between the various genotypes and phenotypes makes genetic counselling and prognostication difficult. 2 Lastly, until recently, there was no specific treatment for this disease that carries a poor prognosis in its severe forms. In 2015, asfotase alfa, a recombinant bone-targeted alkaline phosphatase (ALP) was approved by multiple medicine regulatory authorities, including the European Medicines Agency and United States Food and Drug Administration (US FDA) for use in paediatric-onset HPP. 3
Pathophysiology
There are four isoenzymes of ALP (intestinal, placental, germ cell and TNSAP). TNSAP is found predominantly in the bone, liver and kidney, but also in other cell types. 4 This homodimeric enzyme is located on the extracellular surface of the membrane and hydrolyses phosphates. In HPP, the reduced activity of TNSAP leads to the extracellular accumulation of inorganic pyrophosphate (PPi), pyridoxal 5′-phosphate (PLP) and phosphoethanolamine (PEA). The increased extracellular ratio of PPi/inorganic phosphate (Pi) acts as an inhibitor of skeletal mineralization.1,5 In the normal skeleton, there are two stages of mineralization. The first stage, that remains intact in HPP, involves the formation of hydroxyapatite (HA) crystals from calcium ions (Ca2+) and Pi within the matrix vesicles (MVs). The second stage is the release of HA from the MVs to the extracellular matrix that further elongates with the uptake of extracellular Pi and Ca2+. HA deposits in the collagen fibrils. The increased levels of PPi in HPP inhibit HA propagation (Figure 1).
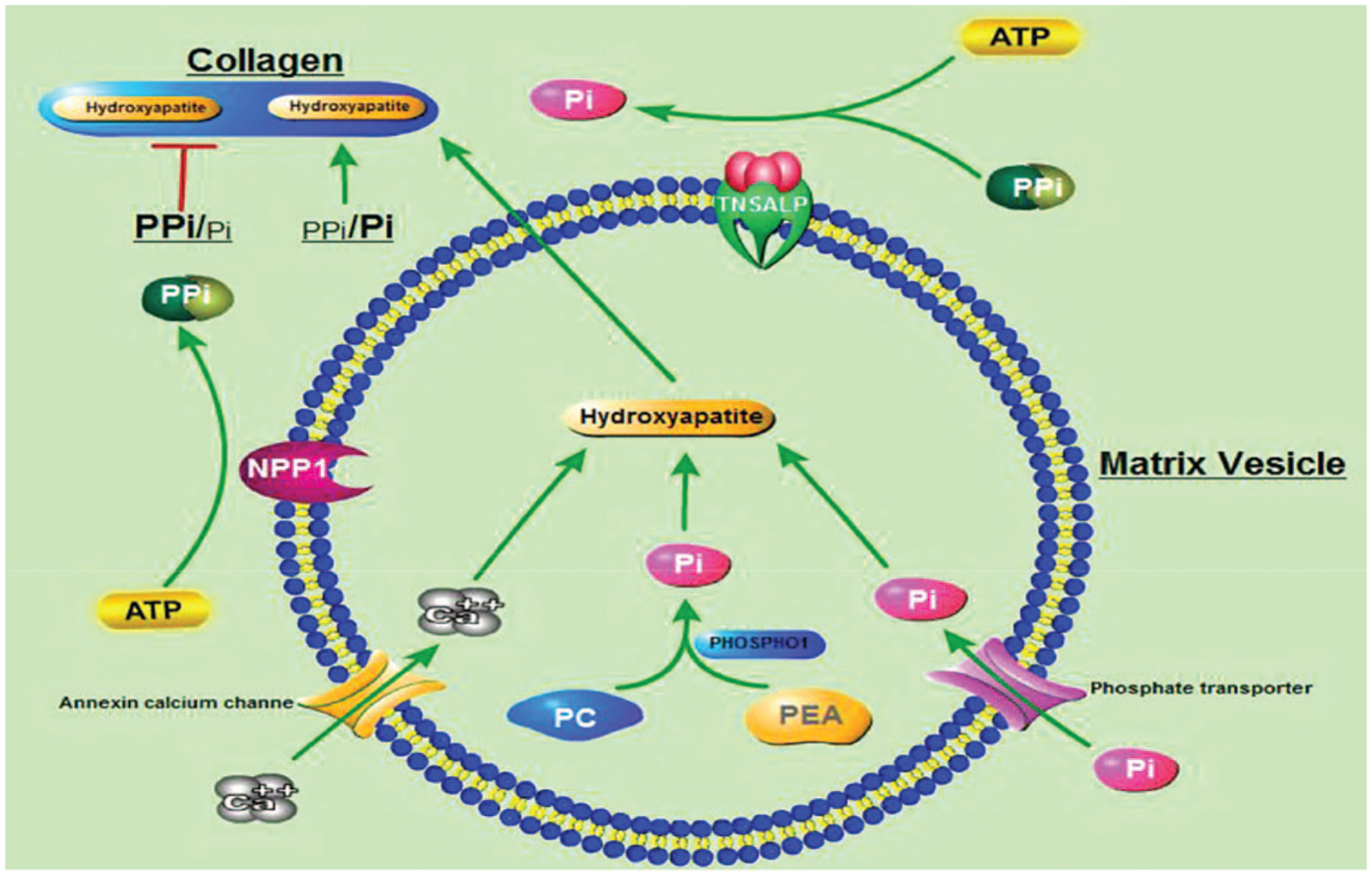
The hypomineralization manifests clinically as a ‘rickets-like’ deformity and bone deformation in infants and children and with a similar clinical picture to osteomalacia once the growth plates close, along with muscle weakness.
One further feature of HPP can be seizures in infants. The deficiency in the dephosphorylation of extracellular PLP is thought to be the cause of seizures in HPP. PLP is a cofactor for the synthesis of important molecules in the central nervous system such as gamma-aminobutyric acid (GABA). However, PLP needs to be dephosphorylated and converted to pyridoxal (PL) in order to enter the cells.1,5 PLP is the active form of B6 and seizures in HPP can respond to the administration of vitamin B6 (pyridoxine).
Epidemiology
The ethnic group with the highest reported incidence of HPP is the Mennonites in Manitoba, Canada. This population is reported to have 1 in 25 individuals carrying a TNSAP mutation and around 1 in 25,000 newborns having lethal HPP. 7 Data from the US suggests that HPP is more prevalent in white people than in black people8–10 and the majority of affected American children have a mild disease.8,11 The prevalence of severe forms of HPP was estimated to be 1 in 100,000 births in Canada. 12 A molecular study has found the prevalence of severe HPP to be 1 in 300,000 in Europe, but the same study pointed that moderate forms of HPP are 50-times more frequent. 13 In addition, two mutations have been reported to cause severe HPP in Japanese patients. 14
Genetics
The alkaline phosphatase-liver (ALPL) gene coding the enzyme TNSAP is located in chromosome 1. 1 HPP can either be autosomal dominantly (AD) inherited or it can be autosomal recessive (AR). The chances of a couple who had one child with perinatal HPP (AR) to have another pregnancy affected is 25%. The penetrance of the dominant mode is not 100% and therefore not all carriers of a dominant mutation will develop the disease. 2 There is also variable expression of the disease within the same family. 15
There are at least 388 mutations of ALPL described. 16 Given the variety of genetic mutations, patients can be compound heterozygotes, which means carriers of two different pathologic alleles, a phenomenon that enhances the phenotypic variety. 17 Some mutations have a dominant negative effect which is seen on heterozygotes with mild or moderate forms of HPP.2,18
Clinical types
There are six different subtypes of HPP as described in Table 1.
Clinical subtypes of HPP.

AD, autosomal dominant; AR, autosomal recessive; CPPD, calcium pyrophosphate dihydrate crystal deposition disease; HPP, hypophosphatasia.
The
The
The
The
The
Diagnosis
Along with recognising the radiographic and clinical characteristics of the disease, the laboratory abnormalities are crucial in making the diagnosis. ALP has different normal levels depending on age of the patient and it is therefore essential to use age-related reference ranges otherwise the diagnosis may be missed. The low levels of ALP that give the name to the disease are not pathognomonic as there may be other causes for reduced ALP. On the other hand, causes for raised ALP such as cholestasis can sometimes mask the disease. The increased plasma PLP levels are considered the most useful finding in association with the ALP, as it is a sensitive and specific test. The plasma and urine PPi levels will also be raised if measured, but these tests are not widely available. The plasma and urine PEA levels are expected to be high but these are not pathognomonic as they can be affected by urine excretion, diet and age. 21
In adult patients presenting with stress fractures and low bone mineral density, HPP can easily be mistaken for osteoporosis. It is essential not to ignore a low ALP measurement in this group and to remember that after a fracture, ALP will be raised and therefore the diagnosis may be missed. If there is clinical suspicion of HPP due to the presentation, then ALP measurements should be repeated once the fracture has healed.
Management
Until recently, the management of HPP has been symptomatic and supportive only. Babies can require ventilation if severely affected, which can be technically difficult. 23 Neonatal seizures can be treated with Vitamin B6 but may become refractory. 24
Management of HPP needs a multidisciplinary team to provide best care to the patient and support to the family. Paediatric patients need input from paediatricians and older patients should be managed by adult physicians with an expertise in metabolic bone diseases (mostly commonly, rheumatologists and endocrinologists). As these cases are rare, care is best provided in specialist centres where teams will be managing more patients with the condition and therefore have more experience. If this is not possible due to geography, then a ‘hub and spoke model’ can be applied where a local team is supported by an expert team from a distance. Orthopaedic surgeons are needed in the team as well as dentists with an interest in the condition. Pain management services are also helpful to help manage pain. Specialist nurses are vital members of the team to provide support, information and help patients to access appropriate care. Psychological support may be required: either in a formal sense via a psychology service or informally by the support of the specialist nurse. Physiotherapy and occupational therapy may also be required.
Patients should be signposted to patient support groups, who can provide information, education and support for those living with HPP. In the US the Soft Bones Foundation (www.softbones.org) and in the UK ‘Soft Bones UK’ (www.softbonesuk.co.uk) can provide support.
Genetic counselling is important for families who may consider having more children. In addition, this is important for patients with HPP as they reach adulthood and may wish to have their own children. At least 388 genetic mutations have been identified and sequencing of the ALPL gene is required. As discussed, the inheritance can be dominant or recessive and there is variable penetrance, so this makes the counselling complex and these discussions are best to be done in a specialist genetics service.
Pain management
Pain is a significant problem in this condition. Involvement of experts in pain management can be helpful and use of anti-inflammatories 25 and neuropathic agents may be advised.
Fracture management
Orthopaedic management of fractures in HPP needs to be different to standard fracture treatment. Femoral fractures or pseudofractures should be rodded to provide stability. Pins and plates should be avoided as the bone is soft and complication rates are high with this approach. 26 In addition to requiring orthopaedic intervention for fractures, patients may need neurosurgery for craniosynostosis. 27
Teriparatide has been reported in case reports to help fracture healing in adults with HPP. Teriparatide is an analogue of the first 34 amino-acids of parathyroid hormone and cannot be used in children. It is licensed for treatment of osteoporosis in postmenopausal women and men at risk of fracture and for glucocorticoid induced osteoporosis. It is given as a 20 µg daily subcutaneous injection and has an anabolic effect on bone.10,28
There is also a case report of six postmenopausal women with HPP treated with teriparatide reducing pain, but no improvement in pain in one premenopausal woman. 29
Avoid bisphosphonates
Bisphosphonates are structural analogues of pyrophosphate and are antiresorptive drugs, which work by their effect on osteoclasts. Bisphosphonates are licensed for osteoporosis, Paget’s disease, malignancy and hypercalcaemia. They are also used off-licence in other bone diseases, including osteogenesis imperfecta and bone marrow oedema syndrome. In HPP, pyrophosphate is one of the mineralization inhibitors that accumulates. Bisphosphonates may worsen the hypomineralization in HPP either directly or by binding zinc or magnesium and compromising any residual TNSAP activity 30 and therefore there is no place for bisphosphonates in the management of HPP. Bisphosphonate-related atypical femoral fracture has been reported. 31
Enzyme replacement therapy
In the 1980s ALP replacement therapy was trialled using infusions soluble ALP from Paget bone disease plasma32,33 or purified from human placenta. 34 These studies showed transient hyperphosphatasia, but there was no clinical or radiographic improvement. There were two case reports of marrow and bone cell transplantation that appeared to rescue two girls with infantile HPP.35,36
Asfotase alfa (Stensiq) is an enzyme replacement therapy that was licensed by the US FDA in 2015 for use in paediatric-onset HPP. Asfotase alfa is a soluble glycoprotein that is composed of two identical polypeptide chains. Each of these chains is made up of the catalytic domain of human TNSAP, the human immunoglobulin G1 Fc domain and a deca-aspartate peptide used as a bone targeting domain.
In an animal model of infantile HPP using TNSAP knockout mice it was shown that the animals remained healthy if given asfotase alfa subcutaneously from birth. 37 This study was followed by trials for infants or young children with perinatal or infantile HPP. A total of 11 patients were recruited into this study. The protocol was to receive one infusion of asfotase alfa intravenously and then three times a week, subcutaneous injections. Overall, one child was withdrawn by the parents due to an infusion reaction and then had skeletal deterioration. A second child died from sepsis, which was unrelated to the treatment. A total of nine children were treated and the results of this study were published in 2012. They showed improvements in muscle strength and skeletal mineralization as well as improved pulmonary, cognitive and motor function. 27 These findings were replicated in a larger cohort of similar patients. 38 At the same time, a study of 13 children who had survive infantile HPP or have severe childhood HPP were studied. 39 They showed rapidly improved skeletal health, improved muscle strength and resolution of pain and disability that persisted after 5 years of treatment. In addition, a cohort of 37 patients with perinatal and infantile forms of HPP was compared with a natural history cohort. The treatment group had been treated for a median of 2.7 years and they showed mineralization of the HPP skeleton including the ribs, improved respiratory function and survival. 40 Asfotase alfa treatment in adolescents and adults with HPP has indicated better mobility. 41
The licensed indication for asfotase alfa is for patients of any age with perinatal/infantile- and juvenile-onset HPP. It is given as a subcutaneous injection at a dose of 6 mg/kg/week administered as 2 mg/kg given three times per week or 1 mg/kg given six times per week. Injection site reactions are common and may limit the tolerability of the six times per week regimen. For perinatal/infantile-onset HPP, the dose may be increased for lack of efficacy (e.g. no improvement in respiratory status, growth, or radiographic findings) up to 9 mg/kg per week administered subcutaneously as 3 mg/kg three time per week.
Reported adverse effects include: injection site reactions (46–90%), lipodystrophy (18–70%), ectopic calcifications (5–55%), vomiting/emesis (3–10%), and systemic hypersensitivity reactions (2–10%).
It is yet to be seen what impact use of asfotase alfa will have in adults with paediatric-onset HPP. Further studies are ongoing and required in this patient group. In the UK, the National Health Service England have commissioned expert centres for the management of HPP. A managed access agreement is in place that allows patients to be treated if they meet certain clinical criteria (Table 2). This is only for patients with a confirmed paediatric onset of disease. Patients have to sign up to the managed access agreement that allows data on their response to be collected. Asfotase alfa treatment can only be initiated and managed via the expert centres. Strict start and stop criteria are laid out and patients have to be regularly assessed to ensure they are only treated if they are benefitting from the treatment. This agreement is currently in place for 5 years and will then be reviewed using the data that has been collected. 42
National Institute for Health and Care Excellence managed access agreement starting criteria for asfotase alfa in the United Kingdom.

HPP, hypophosphatasia.
Asfotase alfa is currently not available in the UK for patients with adult-onset HPP.
Conclusion
HPP remains a complex disease with a variable phenotype. The availability of enzyme replacement therapy is transforming care and outcomes for those patients with perinatal and infantile-onset disease. It is yet to be seen how this treatment will affect outcomes in the longer term and whether it will reduce morbidity from the condition in adulthood. Exciting as this development is, there remain challenges in treating this disease. Asfotase alfa therapy is currently limited to those with the most severe disease. As this is a new treatment, we are not yet aware of any long-term complications of its use. There remains untreated morbidity in those patients who do not qualify for treatment. This continues to require a multidisciplinary team approach. In addition, we do not currently have a licensed treatment for those patients with adult-onset disease. There is ongoing research in this group.
To continue to develop this, it is probably best that patients are managed by the UK National Health Service at one of the commissioned highly specialized centres to allow access to asfotase alfa for those who need it and to allow ongoing research in this group. The future is certainly looking brighter for patients with HPP, but more work is needed.