
Abstract
Human bones are in a continuous process of remodeling that ensures renovation and maintenance of the skeletal mass. Bone remodeling has two phases that are normally coupled and balanced: bone resorption mediated by osteoclasts and bone formation mediated by osteoblasts. An increase in bone resorption over bone formation results in a progressive loss of bone mass and impairment of bone microarchitecture leading to osteoporosis and its associated fractures. Recent advances in the understanding of the molecular and cellular mechanisms involved in the remodeling process have allowed the development of new targets for osteoporosis treatment. Cathepsin K, a cysteine protease, is found in osteoclasts along the bone resorption surfaces and very efficiently degrades type I collagen, the major component of the organic bone matrix. Inhibition of cathepsin K reduces bone resorption but does not impair bone formation particularly at cortical sites. Odanacatib, a potent and highly selective cathepsin K inhibitor, showed prevention of bone loss without reduction of bone formation in preclinical and clinical trials (phase I and II). Odanacatib is currently in a phase III fracture outcome international trial for the treatment of postmenopausal osteoporosis.
Keywords
Introduction
Osteoporosis is a systemic skeletal disease characterized by low bone mineral density and microarchitectural deterioration of bone, leading to skeletal fractures secondary to minimal trauma. Spine, hip and forearm are common sites of those fragility fractures. Owing to the frequent association between fragility fractures and an increase in morbidity and mortality, osteoporosis represents a huge socioeconomic and public health burden for many countries. This chronic disease affects mainly women after the menopause but can also develop in older men. At the age of 50 years, the lifetime fracture risk is 50% for women and 25% for men [Harvey et al. 2010].
In adult life, bone remodeling is the process that ensures renovation and maintenance of bone mass. Bone remodeling is based on the coupled and balanced actions of bone resorption by osteoclasts and bone formation by osteoblasts. An imbalance in this process due to an increase in bone resorption or a decrease in bone formation will lead to continuous loss of bone mass and impairment of bone architecture, ultimately causing fractures.
General measures such as balanced nutrition, moderate exposure to sunlight and regular exercise are important for the development of healthy bones and may retard bone loss in adult life, but are not adequate treatments for osteoporosis. For patients at high fracture risk, treatment with pharmacological agents is indicated to reduce fracture risk. Such drugs are usually categorized as inhibitors of bone resorption (antiresorptives) or stimulators of bone formation (anabolics).
Antiresorptive drugs are very effective in decreasing the risk of vertebral fractures but have a limited effect on nonvertebral fractures. Their action on the remodeling process culminates in a low bone turnover which limits further increases in bone mass. Continuous use may be associated with clinical complications in some patients, including osteonecrosis of the jaw and atypical subtrochanteric fractures.
Anabolic drugs are inducers of osteoblast action. The only anabolic drugs approved for the treatment of osteoporosis are the injectable forms of the recombinant human parathyroid hormone 1-84 or 1-34 (teriparatide). Administration of teriparatide reduces the risk of vertebral and nonvertebral fractures, but its effectiveness may be limited by some factors. The stimulus of bone formation is followed by an increase in bone resorption that blunts the anabolic effect after 18–24 months [Neer et al. 2001]. The cost of this treatment, the requirement of daily injections and concerns about the possible risk of osteosarcoma also limit the acceptance and use of these anabolic agents [Vahle et al. 2002; Subbiah et al. 2010]. In many countries, the use of these drugs is limited to only 24 months
Although significant advances have been made in the last 20 years, there are still unmet needs in the treatment of osteoporosis. Recent research in this area has brought new concepts and the opportunity for the development of new drugs. In this review we focus on a new class of drugs that inhibits the osteoclast reabsorbing enzyme cathepsin K.
Cathepsin K
During the resorption phase of the remodeling cycle, the actions of osteoclasts can be divided into two main types: secretion of H+ creating an acidic environment capable of dissolving the mineral component of the bone (hydroxyapatite); and secretion of proteolytic enzymes capable of digesting the organic matrix (mainly collagen type I). Cathepsin K is the most important of this group of enzymes (Figure 1).
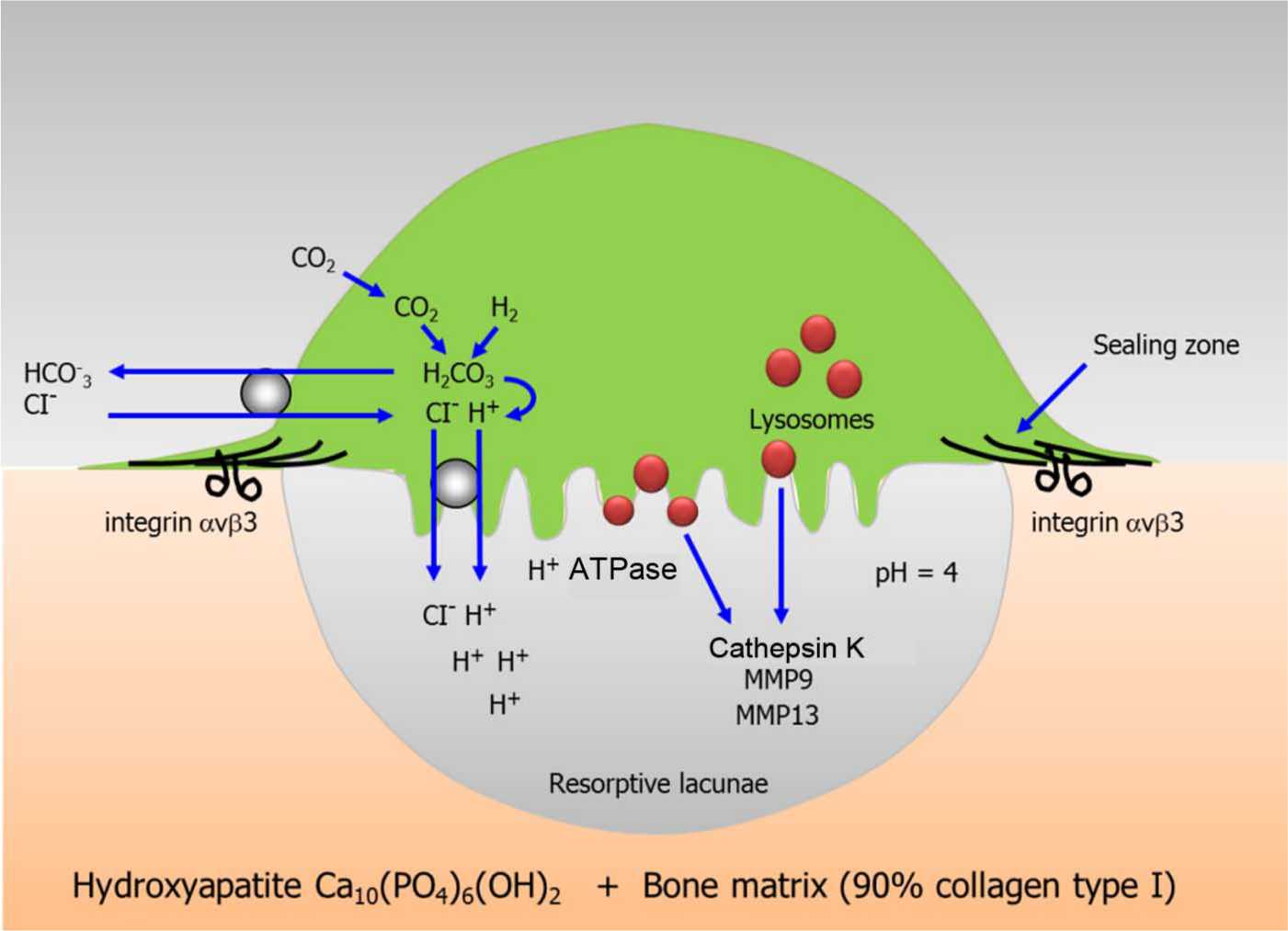
Mechanism of bone resorption by osteoclasts. A sealing zone is created when osteoclasts attach to the bone surface through integrin αVβ3. Secretion of H+ and acidic proteases such as cathepsin K and metalloproteinases (MMP9, MMP13) mediate bone degradation, creating a resorptive lacunae.
Cathepsins are lysosomal proteases found in many types of cells (Figure 2). Approximately a dozen members of this family have been described; these are differentiated by their structure, catalytic mechanism and the kind of proteins they cleave (cysteine, aspartyl or serine proteases). Cathepsin K, a cysteine protease, is found abundantly in osteoclasts along the bone resorption surfaces, in transcytotic vesicles and intracellular lysosomes [Leung et al. 2011; Vaaraniemi et al. 2004]. Cathepsin K is a very efficient collagenase and has a major role in bone matrix degradation necessary for bone resorption. An immature form of cathepsin K (prepro cathepsin K) is catalytically activated to the functional cathepsin K form in low pH environments [Rieman et al. 2001].

Cathepsins: a family of lysosomal proteases. Cathepsins are lysosomal proteases found in many types of cells. They are differentiated by their structure, catalytic mechanism and the kind of proteins they cleave (cysteine, aspartyl or serine proteases). Cathepsin K, a cysteine protease, is abundantly expressed in osteoclasts along the bone resorption surfaces, in transcytotic vesicles and intracellular lysosomes. Cathepsin K has optimal activity at the acidic pH found in lysosomes and efficiently degrades collagen.
During the resorption phase of bone remodeling, cathepsin K is secreted from the osteoclast into the acidic resorption space (pH 4.5) and promotes the degradation of collagen type I, the major component (90%) of the organic bone matrix. Incompletely digested collagen fragments accumulate in the acidic milieu of the intracellular lysosomes where enzymatic hydrolysis by cathepsin K continues. The catalytic action of cathepsin K cleaves the N-telopeptide and the C-terminal telopeptide of type I collagen, generating, respectively, NTx and CTx, two commonly measured resorption biomarkers. Cathepsin K also cleaves collagen type I at multiple sites in triple helical domains and noncollagenous bone matrix proteins such as osteocalcin, osteopontin, ostenectin, proteoglycans and growth factors [Garnero et al. 1998; Bossard et al. 1996; Atley et al. 2000]. Cathepsin K expression in osteoclasts is regulated by the interaction of the receptor activator of nuclear factor kappa B ligand (RANKL) with the cathepsin K gene [Rood et al. 1997; Balkan et al. 2009]. Many inducers of bone resorption and osteoclast differentiation such as tumor necrosis factor (TNF), interleukin 1 (IL-1), IL-6, IL-17, vitamin D and parathyroid hormone may also induce cathepsin K production [Troen, 2006].
Cathepsin K may also be expressed in other cells. Its expression has been found in osteoblasts [Mandelin et al. 2006], osteocytes [Jia et al. 2011], macrophages and smooth muscle cells of the atherosclerotic lesions [Sukhova et al. 1998], synovial fibroblasts and macrophages [Hou et al. 2001], white adipose tissue [Xiao et al. 2006], and in breast and prostate cancer cells [Littlewood-Evans et al. 1997; Brubaker et al. 2003]. This wide distribution may explain the implication of cathepsin K in disorders not related to bone metabolism. Some adverse effects seen with cathepsin K inhibitors in clinical trials may also be explained by this multicellular distribution.
The finding of loss-of-function mutations in the cathepsin K gene, described in a natural occurring disease named pycnodysostosis, showed clearly the relationship between cathepsin K function and bone homeostasis [Gelb et al. 1996]. Pycnodysostosis, a rare autosomal recessive human syndrome of skeletal dysplasia, is associated with short stature, brachycephaly, wide cranial sutures, osteosclerosis, fragility fractures and high bone density. The absence of cathepsin K function leads to a lifelong defect in bone resorption resulting in increased bone mass [Motyckova and Fisher, 2002; Schilling et al. 2007]. The impairment in bone remodeling is reflected by a reduction in bone resorption markers (NTx and CTx). The phenotype observed in pycnodysostosis can be reproduced in the cathepsin K deficient mice by targeted disruption of the cathepsin K gene. These animals develop an osteopetrotic skeleton characterized by increased trabecular and cortical bone mass due to a diminished bone resorption. Histological examination of their bones shows a normal rate of mineralization, normal or increased osteoclast numbers, but a decrease in the osteoclast ability to resorb bone matrix [Gowen et al. 1999]. The observation that cathepsin K knockout mice [cathepsin K (-/-)] have more osteoclasts was clarified by a study indicating that cathepsin K may play an important role in osteoclast apoptosis and senescence [Chen et al. 2007]. Although pycnodysostosis patients may be at a higher risk for fracture, cathepsin K (-/-) mice did not show poor bone quality. Cathepsin K (-/-) mice have a high rate of bone formation and increased bone strength at the vertebral body and femoral midshaft compared with normal animals [Pennypacker et al. 2009]. The changes may be explained by increases in local bone growth factors due to their decreased clearance in the absence of cathepsin K [Fuller et al. 2008].
Cathepsin K inhibitors in preclinical studies
The importance of cathepsin K in bone matrix degradation makes it a suitable pharmaceutical target for the treatment of osteoporosis and other diseases associated with high bone turnover. Reversible inhibitors of the active form of human cathepsin K have been developed. The inhibitors cannot be evaluated in rodent species due to the lack of amino acid homology between rodent and human cathepsin K [Rodan and Duong, 2008]. Because the structures of cathepsin K in nonhuman primates, rabbits and humans are almost identical, those animals have been used selected for studying the cathepsin K inhibitors in vivo [Rodan and Duong, 2008].
The cathepsin K inhibitors relacatib, balicatib, ONO-5443 and odanacatib were evaluated in preclinical and clinical studies. In a 9-month study, treatment of ovariectomized monkeys with relacatib, an inhibitor of human cathepsins K, L and V, showed a marked, dose-dependent reduction in bone resorption markers, serum CTx and urinary NTx. Relacatib also preserved the areal bone mineral density (BMD) in these animals [Stroup et al. 2009]. Similar results were obtained in an 18-month study with balicatib, a specific cathepsin K inhibitor [Jerome et al. 2011]. In both studies, histomorphometric analysis demonstrated that these agents reduced indices of bone resorption at trabecular and cortical sites [Stroup et al. 2009; Masarachia et al. 2011]. ONO-5334 decreased serum and urinary CTx levels without affecting bone formation in ovariectomized monkeys [Ochi et al. 2011].
Odanacatib, a more potent and selective cathepsin K inhibitor, was evaluated in ovariectomized rabbits and monkeys [Masarachia et al. 2011; Pennypacker et al. 2011]. After treatment with odanacatib, these estrogen-deficient models showed gains in BMD and reduction in serum CTx and urinary NTx [Masarachia et al. 2011; Pennypacker et al. 2011]. Histomorphometry and analysis of serum tartrate resistant acid phosphatase 5b (TRAP5b) levels showed a marked elevation of the osteoclast numbers in the odanacatib-treated animals compared with those vehicle-treated [Masarachia et al. 2011; Pennypacker et al. 2011]. In another study, ovariectomized rabbits were divided into three groups of treatment: odanacatib; L-235 (a lesser selective inhibitor of cathepsin K); or alendronate. Although all treatments showed prevention of bone loss, the cathepsin K inhibitors did not reduce bone formation rate as observed with alendronate [Pennypacker et al. 2011].
In ovariectomized monkeys, treatment with relacatib, balicatib and odanacatib reduced trabecular bone remodeling like other resorption inhibitors, but endocortical bone formation was maintained [Stroup et al. 2009; Masarachia et al. 2011; Pennypacker et al. 2011]. Odanacatib and balicatib stimulated periostal bone formation in the femur, and increased cortical bone thickness and volume and bone strength in the hip region of ovariectomized monkeys [Jerome et al. 2011; Cusick et al. 2011]. Cathepsin K inhibition had no effect on osteoclastogenesis or survival of osteroclasts [Leung et al. 2011]. Osteoclasts isolated from cathepsin K (-/-) mice or treated with cathepsin K inhibitors retain normal cell development and functions including differentiation, migration, polarization, activation and secretion In contrast to untreated cells that produce deep trail-like resorption lacunae, osteoclasts treated with odanacatib form small discrete shallow pits. Intracellular vesicles with high concentrations of cathepsin K and collagen fragments appear within osteoclasts exposed to odanacatib.
Based on the encouraging results of preclinical studies, several cathepsin K inhibitors have advanced into clinical development.
Clinical pharmacology
Cathepsin K inhibitors have pharmacokinetic properties very different from other antiresorptive drugs such as bisphosphonates or denosumab [Cremers et al. 2005; Body et al. 2006]. They are small molecules with good oral absorption showing a combination of potency and metabolic stability. Oral bioavailability of cathepsin K inhibitors depends on formulation and is higher than oral bisphosphonate bioavailability [Kassahun et al. 2011; Yamashita et al. 2006]. Cathepsin K inhibitors do not need to bind to bone to exert their activities and are not sequestered in bone tissue. It is their concentration in the resorption lacunae and in the cell that is relevant to their action. Cathepsin K inhibitors are metabolized by cytochrome enzymes and may interact with other drugs [Isabel et al. 2010], but there is no concern about fasting or food effects.
Cathepsin K inhibitors in clinical trials
Relacatib (SB-462795) is an oral compound developed by GlaxoSmithKline which acts nonselectively on cathepsins K, L and V. Clinical trials with this drug were discontinued after phase I because of drug–drug interactions with the commonly prescribed medications acetaminophen, ibuprofen and atorvastatin [Kumar et al. 2007; Podgorski, 2009].
Balicatib (AAE-581, Novartis) is a highly selective nitrile-based cathepsin K inhibitor in enzyme-based assays, but is not so selective in whole cell assays due its high concentration in lysosomes, a property called lysosomotropism [Falgueyret et al. 2005; Black and Percival, 2006]. Phase II trials were conducted in subjects with osteoarthritis and osteopenia/osteoporosis. The trial designed to evaluate balicatib in low bone mass patients enrolled 675 postmenopausal women in a 1-year ranging study, comparing four daily oral doses of the drug (5, 10, 25 or 50 mg) with placebo. Treatment with balicatib 25 and 50 mg reduced bone resorption markers (CTx 61%, urinary NTx 55%) without change in bone formation markers [Vasiljeva et al. 2007; Costa et al. 2011]. Balicatib treatment was also associated with a dose-related increase in BMD, reaching 4.5% at the lumbar spine and 2.2% at the total hip with the 50 mg weekly dose. Despite these results, its clinical development was discontinued due to skin-related adverse events including rashes and scleroderma-like lesion similar to morphea [Rünger et al. 2012]. The dermatologic toxicities might be due to the lysosomotropism described above. The high concentration of balicatib in the lysosomes may potentially inhibit cathepsins B, L and S expressed in skin fibroblasts, leading to a reduction of dermal matrix turnover [Falgueyret et al. 2005].
ONO-5334 (ONO Pharmaceutical) is a potent hydrazine-based nonlysosomotropic inhibitor of cathepsin K [Ochi et al. 2011]. A multicenter, randomized, double-blind trial enrolled 285 postmenopausal women with osteoporosis randomized to receive placebo, alendronate 70 mg weekly, or one of three regimens of oral ONO-5334 (50 mg twice daily, 100 mg once daily or 300 mg once daily) [Eastell et al. 2011]. At 12 months, ONO-5334 decreased serum and urinary CTx and urinary NTx levels with little or no reduction of bone formation markers compared with alendronate. The 300 mg dose inhibited bone-resorption markers comparable to alendronate and only this dose significantly decreased the levels of the bone formation markers bone-specific alkaline phosphatase (BSAP) and procollagen type 1 N-terminal propeptide (P1NP). Levels of tartarate-resistant acid phosphatase isoform 5b (TRAP5b), a marker of osteoclast number, were suppressed by alendronate but increased by the cathepsin K inhibitor. After 1 year, the 300 mg dose of ONO-5334 achieved significant increases in the BMD of the lumbar spine (5.1%), total hip (3%) and femoral neck (2.6%) compared with placebo. Those BMD increases were comparable to the alendronate results. There were no clinically relevant safety concerns during the study. No difference was detected in the rate of skin adverse events among all study groups. This compound is no longer under clinical development for the treatment of osteoporosis.
Odanacatib (MK-822) is a nonbasic, nonlysosomotropic, nitrile-based molecule and a highly selective cathepsin K inhibitor under development by Merck [Podgorski, 2009; Gauthier et al. 2008; Desmarais et al. 2009] (Figure 3). Two phase I clinical trials enrolled healthy postmenopausal women short courses of odanacatib in daily (n = 30) and weekly (n = 49) dosing regimens. A half-life of 66–93 hours was observed, allowing for once weekly dosing [Stoch et al. 2009]. After 21 days of treatment, serum CTx and urinary NTx were decreased from baseline by 62% of baseline levels at weekly doses of ≥50 mg. Odanacatib was well tolerated and no serious adverse events were reported. A phase II randomized, multicenter, placebo-controlled trial enrolled 399 postmenopausal women with low BMD (T-score ≤-2.0 and ≥-3.5) allocated to one of the following weekly odanacatib treatments: 3, 10, 25 and 50 mg [Bone et al. 2011a]. The trial was designed as a 12-month study with a 12-month extension period. The primary endpoint was the percentage change in lumbar spine BMD from baseline at month 12. Secondary endpoints included changes in BMD at other sites and evaluation of bone turnover markers. After 12 months of treatment, dose-dependent increases at the lumbar spine and femoral neck were observed. At the highest weekly dose (50 mg), BMD at the lumbar spine and femoral neck increased by 3.4% and 2.5%, respectively. Further gains were seen at 24 months: 5.7% increase in lumbar spine BMD versus placebo and 4.1% in the total hip. Urinary NTx was reduced by 58% versus placebo after 12 months of treatment. During this period, the odanacatib 50 mg weekly dose induced only a small reduction in the levels of bone formation markers (20% for BSAP and 30% for P1NP). After 24 months, the urinary NTx remained reduced while bone formation markers levels gradually tended to return toward baseline. Histomorphometry performed in biopsies of 32 patients showed no remarkable differences in the activation frequency and bone formation rate between odanacatib-treated and placebo-treated patients. During this trial all doses were well tolerated; adverse events were similar in the treatment and placebo groups [Bone et al. 2011]. Because of the experience in the balicatib study of possible off-target effects, particular attention was paid to skin and pulmonary events, and no signals were observed.

Chemical structure of odanacatib (MK-0822).
In an extension of the phase II study, 189 patients of the original 280 subjects participated [Eisman et al. 2011]. During that study, participants treated with odanacatib 50 mg for the entire 3 years increased lumbar spine BMD by 7.9%, total hip BMD by 5.8%, femoral neck BMD by 5% and trochanter BMD by 7.4% from baseline values. Participants treated continuously showed a lowering of 50% in urinary NTx from baseline values compared with a 17% decline for those treated with placebo for the 3-year period. Levels of bone formation markers P1NP and BSAP fell in the initial months of treatment, reaching the lowest level at month 6. At the end of the 3-year extension study, BSAP levels were 18% lower than baseline levels and P1NP levels had returned to baseline levels. TRAP5b levels remained above baseline during the 3 years of odanacatib treatment, but were not significantly different from those in the placebo group. Subjects who discontinued odanacatib after 2 years of therapy experienced bone loss at all sites, which was faster than during the first 6 months of placebo (Figure 4). BMD returned to or near baseline levels within 12 months of stopping odanacatib. Within a month after switching from active treatment to placebo, bone turnover markers increased above baseline and then returned to baseline values by the end of the third year. With the exception of a greater number of noncomplicated urinary tract infections in the odanacatib group (n = 12) compared with placebo (n = 3), the overall adverse events were similar in all treatment groups, including skin events.

Mean percentage change from baseline over time for the full-analysis-set population (LOCF) in lumbar spine BMD (primary endpoint) (a) and femoral neck BMD (b) at 5 years for three of the randomization groups.
The 5-year extension results of the phase II trial were reported recently [Langdahl et al. 2012]. Participants who received odanacatib 50 mg once a week continuously for 5 years showed almost linear increases in BMD from baseline at the lumbar spine (11.9%), total hip (8.5%) and femoral neck (9.5%) (Figure 4). The serum levels of BSAP remained close to baseline while the level of urinary NTx was decreased by 67.4%. At 5 years, levels of TRAP5b in the odanacatib treatment group were similar to those in the placebo group, demonstrating maintenance of osteoclast survival despite the persistent decrease in bone resorption. The phase II trial has been extended to 10-years to further assess the effects on safety and efficacy of the odanacatib treatment in osteoporotic postmenopausal women.
Four recent studies have extended these observations. A double-blind, randomized, multicenter study evaluated the efficacy and safety of 3 once-weekly doses of odanacatib for 52 weeks in Japanese women and men (4%) with low bone mass [Nakamura et al. 2012]. The subjects (n = 287) were randomly assigned to take placebo (n = 73) or odanacatib 10 mg (n = 74), 25 mg (n = 71) or 50 mg (n = 69). After 52 weeks, the odanacatib 50 mg group showed a lumbar spine BMD increase of 5.9% compared with a placebo increase of 0.5% and a total hip BMD increase of 2.7% compared with a placebo decrease of 0.4%. Odanacatib reduced bone markers of resorption in a dose-dependent manner and was well tolerated in Japanese patients
An imaging study using quantitative computed tomography and finite element analysis evaluated the impact of odanacatib on trabecular and cortical bone compartments. After 2 years of treatment, odanacatib increased areal and volumetric BMD and bone strength in the hip and spine [Brixen et al. 2012].
The effect of odanacatib on cortical geometry and bone strength was further evaluated in the same study described above, which also used high resolution quantitative computerized tomography (HRpQCt) as an exploratory endpoint. This randomized, double-blind placebo-controlled trial using HRpQCt of the distal radius and distal tibia included a total of 214 postmenopausal women (n = 214, average age 64 years) with low bone mass were randomized to oral odanacatib 50 mg or placebo once weekly for 2 years [DePapp et al. 2012]. Odanacatib therapy was associated with greater increases in lumbar spine areal BMD at 12 months (3.49% treatment difference, p < 0.001). After 2 years, odanacatib increased cortical and trabecular volumetric density, improved cortical thickness of the distal radius and distal tibia, and improved the estimated bone strength in the distal radius compared with placebo.
The effects of odanacatib 50 mg once weekly on BMD and biochemical markers of bone turnover were evaluated over 24 months in patients previously treated with alendronate for ≥3 years. This randomized, double-blind, placebo-controlled, 24-month study enrolled 243 postmenopausal women ≥60 years of age with low BMD T-score (range ≤-2.5 but >-3.5) at the total hip, femoral neck or trochanter without prior hip fracture [De Villiers et al. 2012]. In the odanacatib group, significant BMD changes from baseline at 24 months compared with placebo were seen in the femoral neck (1.73%), trochanter (1.83%), total hip (0.83%) and lumbar spine (2.28%) (Figure 5). Markers of bone turnover increased in the subjects switched from alendronate to placebo. The increase in urinary NTx was less and the increases in biomarkers of bone formation, P1NP and BSAP were greater with odanacatib than with placebo. The overall safety profile appeared similar between odanacatib 50 mg once weekly and placebo.
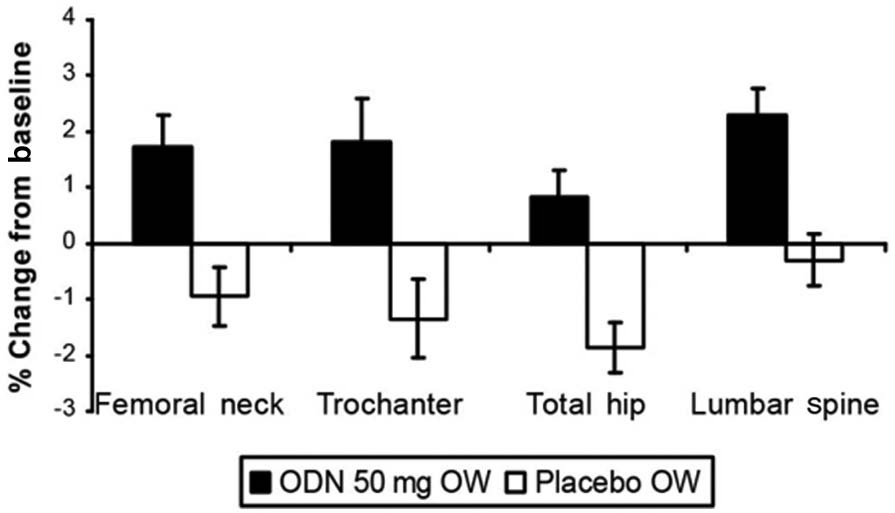
Effects of odanacatib versus placebo on percentage change from baseline in BMD (least square mean ± standard error) at 24 months in postmenopausal women previously treated with alendronate.
An international multicenter placebo-controlled phase III fracture outcome trial enrolled more than 16,000 postmenopausal women from 387 research centers in 40 countries taking 50 mg odanacatib once weekly in a double-blind treatment. In June 2012, based on the first preplanned interim analysis of this event-driven trial, the independent external Data Monitoring Committee (DMC) recommended early closeout of this study due to robust efficacy and a favorable benefit/risk profile [Reuters, 2012]. The DMC noted that safety issues remain in certain selected areas. The study is being closed out. A blinded placebo controlled extension of the trial is underway and will allow further safety and efficacy monitoring for a total of 5 years. Data from this extension trial will be included in the regulatory filings of odanacatib, which are now anticipated to occur in 2014.
Summary
Inhibition of cathepsin K provides a unique mechanism of altering bone metabolism to treat osteoporosis. Current antiresorptive agents such as bisphosphonates and denosumab decreased the number and/or function of osteoclasts, resulting in decreased bone resorption and secondary inhibition of bone formation. Cathepsin K inhibitors, in contrast, do not decrease (and may increase) the number of osteoclasts present in bone tissue. These osteoclasts have impaired capacity to resorb bone matrix but they appear to remain capable of cytokine signaling that preserves osteoblast activity [Baron et al. 2011]. Although cathepsin K inhibitors have less potent effects on decreasing bone resorption than do bisphosphonates and denosumab, a more positive bone balance is achieved because bone formation is preserved over time. This likely explains the progressive increase in BMD observed during 5 years of continuous therapy with odanacatib [Eisman et al. 2011].
Preclinical data document improved bone mass and a unique effect on increasing cortical thickness and long bone strength with odanacatib. If these effects occur in humans, treatment may result in improved protection from nonvertebral fractures compared with conventional therapies. Results of the phase III fracture trial, which are anticipated in 2014, will then be of special interest.
The safety profile of cathepsin K inhibitors may also be different from current treatments. Concerns about ‘over suppression’ of bone turnover which may result in osteonecrosis of the jaw (ONJ) and atypical fracture may be less than with current potent antiresorptive agents. However, off-target inhibition of other cathepsins or of cathepsin K in nonskeletal tissues poses a different worry. This was a large enough problem to halt the development of some cathepsin K inhibitors. The large phase III placebo-controlled study with odanacatib will also provide important insight regarding safety and tolerability.
If the ability of odanacatib to reduce fracture risk is confirmed and provided important safety concerns are not found, this agent will be a welcome addition to our treatment options. It would be an appropriate first-line agent but would be particularly attractive as an option for patients with intolerance or contraindications (e.g. renal insufficiency) to bisphosphonates. It may also be an appealing drug to use after 3–5 years of bisphosphonate or denosumab therapy when concern about long-term inhibition of bone remodeling may arise.
Conclusion
The inhibition of cathepsin K provides a novel mechanism of modulating bone remodeling. Preclinical and early clinical data support the potential effectiveness of these agents as treatments for osteoporosis.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Dr Zerbini has received research grants, Consulting fees and/or honorarium from Pfizer, Sanofi, GSK, Amgen Lilly, Merck, Aché and Servier. Dr McClung has received research grants, consulting fees and/or honorarium from Amgen, Lilly, Merck, Novartis and Warner-Chilcott.