
Abstract
Background:
The current standard-of-care chemotherapy for treatment-naïve epithelial ovarian cancer is paclitaxel plus carboplatin.
Objectives:
To compare the efficacy and safety of pegylated liposomal doxorubicin (PLD) plus carboplatin versus paclitaxel plus carboplatin as first-line treatment in patients with epithelial ovarian cancer.
Design:
This was an investigator-initiated, multicenter, open-label, randomized, noninferiority trial.
Methods:
This trial with a prespecified noninferiority margin (HR0) of 1.2, was conducted in 20 clinic centers in China. Eligible patients were randomly assigned in a 1:1 ratio to receive either PLD (30 mg/m2 on day 1) plus carboplatin (area under the curve (AUC) 5 on day 1; experimental group) or paclitaxel (175 mg/m2 on day 1) plus carboplatin (AUC 5 on day 1; control group) for up to six cycles. The primary endpoint was progression-free survival (PFS). The key secondary endpoints included overall survival (OS), objective response rate (ORR), disease control rate (DCR), and safety.
Results:
Between March 21, 2019, and December 8, 2021, 395 eligible patients were enrolled, of whom 195 were randomly assigned to receive PLD-carboplatin and 196 received paclitaxel-carboplatin. Median PFS was 35.3 months (95% confidence interval (CI), 23.7–46.9) in the experimental group and 35.0 months (95% CI, 26.9–43.1) in the control group (hazard ratio = 0.99, 95% CI, 0.73–1.35; p = 0.94). There were no statistically significant differences between the two groups in median ORR (80.5% vs 79.2%), DCR (90.2% vs 83.3%), 2-year OS rate (96.2% vs 92.4%), or 4-year OS rate (87.6% vs 82.4%; all p > 0.05). In the experimental and control groups, 165 (84.6%) and 169 (86.2%) patients experienced at least one adverse event (AEs). Alopecia (9.2% vs 28.1%, p < 0.001), peripheral sensory neuropathy (2.1% vs 17.9%, p < 0.001), and febrile neutropenia (1.0% vs 6.1%, p = 0.01) were less common in the PLD-carboplatin group compared to the paclitaxel-carboplatin group.
Conclusion:
The superior tolerability and comparable efficacy of PLD plus carboplatin over paclitaxel plus carboplatin as a first-line treatment for epithelial ovarian cancer suggest that substituting paclitaxel with PLD is both feasible and potentially more beneficial.
Trial registration:
This trial is registered with ClinicalTrials.gov, NCT03794778.
Keywords
Introduction
Globally, epithelial ovarian cancer was the eighth most common cancer in women, accounting for an estimated 3.4% of cases and 4.8% of cancer deaths in 2022. 1 The absence of effective screening and early diagnostic strategies resulted in approximately 70.0% of patients being diagnosed with advanced-stage disease, 2 posing significant challenges to effective treatment and survival. Currently, standard-of-care therapy for newly diagnosed disease typically involves a combination of cytoreductive surgery and platinum-based systemic chemotherapy, with paclitaxel and carboplatin being the preferred standard regimen in the first-line setting. 3 However, this treatment regimen significantly impacted the quality of life due to frequent toxicities, including alopecia and neurotoxicity.4,5 Consequently, there is a compelling imperative to explore and develop more efficacious alternative strategies.
Considering epithelial ovarian cancer’s tendency to present at advanced stages, treatment options were typically constrained by the balance between therapeutic benefits and toxicities. The emergence of liposomal drug-delivery systems had introduced a compelling and viable strategy to mitigate toxicity and enhance treatment efficacy, thereby demonstrating potential advantages in clinical outcomes for patients.6,7 Pegylated liposomal doxorubicin (PLD; Caelyx®) represented an improved formulation of conventional doxorubicin, with reduced cardiotoxicity and an improved pharmacokinetic profile. Substantiated by the robust evidence garnered from several phase III clinical trials, PLD had established itself as a valuable therapeutic modality in the arsenal against a diverse array of malignancies.8–10 In addition, PLD was approved by the Food and Drug Administration and European Medicines Evaluation Agency as a single agent for the treatment of advanced epithelial ovarian cancer patients failing first-line platinum-based treatment. 11 Subsequent randomized phase III trials compared PLD combination regimens in recurrent epithelial ovarian cancer, which showed significantly longer progression-free survival (PFS) with a PLD plus carboplatin versus paclitaxel plus carboplatin, without impairing quality of life.12,13 In newly diagnosed disease, the randomized phase III MITO-2 trial 14 suggested that although PLD plus carboplatin did not significantly prolong PFS compared to the standard therapy-paclitaxel plus carboplatin, it showed a favorable safety profile and was recommended by the National Comprehensive Cancer Network as an “Other Recommended Regimen” for first-line chemotherapy. 3
Duomeisu®, as a Chinese-developed PLD, boasted a distinctive liposomal structure coated with methoxypolyethylene glycol that optimally refines the pharmacokinetic properties of the drug, enhancing its accumulation within the tumor microenvironment while concurrently alleviating adverse events (AEs) such as cardiotoxicity. Duomeisu® and the reference drug Caelyx® have completed bioequivalence studies and have been approved by the National Medical Products Administration of the People’s Republic of China for passing the consistency evaluation of generic drug quality and efficacy. 15 The results of the relevant bioequivalence trials have been publicly published. Studies have confirmed that the two products are highly consistent in terms of active pharmaceutical ingredients, liposomal structure, and pharmacological mechanism of action. Numerous studies have substantiated Duomeisu® safety and efficacy in the treatment of various solid tumors, including epithelial ovarian cancer.16–18 Nevertheless, there remains a paucity of clinical data comparing the PLD (Duomeisu®) with paclitaxel when either was combined with carboplatin for patients with epithelial ovarian cancer in the first-line setting. This study aims to further provide clinical usage data of PLD in the Chinese population, thereby enriching the localized evidence-based medical data.
In this scenario, we conducted this investigator-initiated multicenter, randomized, noninferiority trial to assess the safety and efficacy of PLD plus carboplatin compared with paclitaxel plus carboplatin as the first-line treatment for epithelial ovarian cancer in China. This trial promised to fill the gap of PLD for the first-line treatment of epithelial ovarian cancer in China, potentially offering a more personalized, effective, and less toxic therapeutic option.
Methods
Study design and participants
This was a multicenter, randomized, parallel, open-label trial conducted at 20 sites in China (Supplemental Appendix 1). Notably, the design of this study followed the rigorous methodology characteristic of phase III trials. Eligible patients had newly diagnosed International Federation of Gynecology and Obstetrics (FIGO) stage IC–IV epithelial ovarian, primary peritoneal, or fallopian tube cancer, confirmed by histopathological/cytopathology evaluation. Patients had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, or cancer antigen-125 (CA-125) assessable disease according to Gynecologic Cancer Inter Group (GCIG) criteria. Additional inclusion criteria included age between 18 and 75 years old, Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–2, life expectancy of more than 3 months, adequate coagulation parameters, and adequate bone marrow, liver, and renal function. Key exclusion criteria included low malignant potential ovarian tumors, previous chemotherapy or abdominopelvic radiotherapy, planned abdominal or pelvic chemotherapy, known allergy to macromolecular protein preparations or any component of study drugs, or a history of grade ⩾2 sensory or motor neuropathy. Full eligibility criteria are provided in the Supplemental Appendix 2.
The study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines and was approved by the ethics committee of all participating centers (Approved No. 20180148, etc. in Supplemental Appendix 1). All patients provided written informed consent before enrollment. The trial is registered with ClinicalTrials.gov, number NCT03794778.
Randomization
Eligible patients were randomly assigned to receive PLD plus carboplatin (experimental group) or paclitaxel plus carboplatin (control group) in a 1:1 ratio, using a prepared computerized block design, stratified according to the chemotherapy (adjuvant vs neoadjuvant), FIGO stage (IC, II, III, or IV), and ECOG PS (0, 1, or 2). The assigned patient number of each patient was documented by using an enrollment log and participant identification number list. The PLD used in the experimental group of this study was Duomeisu®, manufactured by CSPC Ouyi Pharmaceutical Co., Ltd, Shijiazhuang, China.
Procedure
Eligible patients were randomly assigned to receive PLD (30 mg/m2 on day 1) plus carboplatin (area under the curve (AUC) 5 on day 1; experimental group) or paclitaxel (175 mg/m2 on day 1) plus carboplatin (AUC 5 on day 1; control group) for up to six cycles (stage IC disease, 3–6 cycles; stages II–IV disease, six cycles) as neoadjuvant or adjuvant therapy. In both groups, treatment was initially administered for three cycles, and patients with stable disease (SD), complete response (CR), or partial response (PR) continued treatment for the remaining cycles. Patients with advanced-stage disease who are not suitable for primary debulking surgery (PDS) due to advanced age, frailty, poor PS, comorbidities, or disease that is unlikely to be optimally cytoreduced would receive neoadjuvant chemotherapy. Besides, patients who could not achieve R0 interval debulking surgery after multidisciplinary team discussion would also receive neoadjuvant therapy, while others would receive adjuvant systemic chemotherapy after PDS. Those undergoing neoadjuvant chemotherapy received 2–4 cycles of chemotherapy before cytoreductive surgery, with the remaining chemotherapy starting within 6 weeks after cytoreductive surgery, following the same chemotherapy regimen as the initial chemotherapy regimen, for a total of 6 cycles. For patients who have completed six cycles of treatment and judged by the investigator to be feasible to continue treatment with this regimen, treatment may be continued until disease progression or intolerable toxicity occurs, whichever occurs first, up to a maximum total of eight cycles. The experimental and control groups were pretreated with glucocorticoids before the initial administration of the drug.
Assessments
Radiological response was assessed by the investigator per RECIST version 1.1, using magnetic resonance imaging or computed tomography initially at baseline, then every three cycles during the treatment period. GCIG CA-125 response was defined as at least a 50% reduction in CA-125 levels from a pretreatment sample, which must be maintained for at least 4 weeks. CA-125 testing was performed at baseline, then every cycle during study treatment. The method used at screening was used for each subsequent tumor assessment.
In this study, all reported AEs were assessed by the investigators. Safety was assessed through the monitoring of AEs, laboratory testing, measurement of vital signs, and physical examination from the first dose of the study drug until 30 days after the last dose. The severity of AEs was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
Endpoints
The primary endpoint was PFS, defined as the interval between randomization and documented disease progression or death from any cause.
The secondary endpoints included overall survival (OS), objective response rate (ORR), disease control rate (DCR), and safety. OS was calculated the time from the randomization date to the date of death due to any cause. ORR was defined as the proportion of patients with at least one target lesion who achieved a CR or PR. DCR referred to the proportion of patients with at least one target lesion who achieved CR, PR, or SD.
Statistical analysis
Based on the results of previous studies, 14 the median PFS times were 19.0 and 16.8 months with PLD plus carboplatin and paclitaxel plus carboplatin, respectively. Assuming a one-sided significance level of 2.5% and a statistical power of 85%, at least 132 PFS events are needed to show noninferiority of the experimental group against the control group (expected hazard ratio (HR), 0.85) with a noninferiority margin (HR0) of 1.2. To observe this number of events, at least 392 participants need to be enrolled (196 per group), considering a projected dropout rate of 15%.
The full analysis set (FAS), which strictly followed intent to treat analysis, consisted of all randomized patients who received at least one dose of study drugs. Patients in the FAS who met all the trial criteria, complied with the protocol requirements, and had no major protocol deviations, were included in the per-protocol set (PPS). Efficacy analyses were performed in the FAS and PPS. Safety was assessed in the safety set (SS), which was defined as all patients who received at least one dose of study drugs after randomization and had safety records.
Categorical data were presented by numbers and percentages, and continuous data were expressed as mean with standard deviation, median with 95% confidence interval (CI), or median with range. For categorical data, the Chi-square test or Fisher’s exact test were used for between-group comparisons. For continuous data, between-group comparisons were analyzed with independent samples t tests or Mann–Whitney U test. Median PFS and OS were estimated using Kaplan–Meier estimates with corresponding 95% CIs. HRs were assessed using a Cox regression analysis with corresponding 95% CIs for the HR point estimate. To show the consistency of the treatment effect in subgroups based on stratification factors and patients’ characteristics, a PFS analysis was performed in which the HR and 95% CI were calculated with the use of a Cox model. Besides, univariate and multivariate analyses with Cox proportional hazards model were carried out to identify the independent factors. Statistical analyses were performed using SPSS 27.0 (IBM Corporation, Armonk, NY, USA). All statistical tests were two-sided, with significance set at p < 0.05.
In accordance with the journal’s guidelines, we will provide our data for independent analysis by a selected team by the Editorial Team for the purposes of additional data analysis or for the reproducibility of this study in other centers if such is requested. Moreover, this study adhered to the CONSORT 2025 statement: Updated guideline for reporting randomized trials 19 and the completed checklist is available as Supplemental Appendix 3.
Results
Patient characteristics
Between March 21, 2019, and December 8, 2021, of the 396 patients screened, 395 were randomized and 391 (experimental group, n = 195; control group, n = 196) were included in the FAS and SS (Figure 1). The reasons for four patients not being included in the FAS and SS were not receiving the allocated intervention (experimental group, n = 2; control group, n = 1) and data missing (control group, n = 1). Additionally, 90 patients were excluded from the PPS owing to not completing 3–6 cycles of treatment. Correspondingly, a total of 301 patients (experimental group, n = 142; control group, n = 159) comprised the PPS population.

Trial profile.
The baseline characteristics were well balanced between the trial groups (Table 1). Patients in the experimental group and control groups had a median age of 55.0 years (range, 29.0–75.0) and 56.0 years (range, 30.0–75.0), respectively. Most enrolled patients in the experimental and control groups were diagnosed with epithelial ovarian cancer (94.4% vs 89.8%), had FIGO stage III disease (62.1% vs 59.2%), and had ECOG PS of 0–1 (86.2% vs 89.3%).
Characteristics of patients at baseline.

Residual disease residual lesions were identified and documented following the surgery. Data are n (%) or median (range).
BRCA, breast cancer gene; CA-125, cancer antigen-125; ECOG, Eastern Cooperative Oncology Group; FIGO, International Federation of Gynecology and Obstetrics.
Treatment
As of the data cutoff (July 30, 2024), the median duration of study follow-up was 34.2 months (95% CI, 29.8–36.8) in the experimental group and 30.3 months (95% CI, 27.2–37.1) in the control group. The median number of exposure cycles was 6 (range, 1–8) for the experimental group and control group.
Of the FAS population, 142 (72.8%; 26 IC disease complete ⩾3 cycles and 116 II–IV disease complete ⩾6 cycles) of 195 patients in the experimental group and 159 (81.1%; 27 IC disease complete ⩾3 cycles and 132 II–IV disease complete ⩾6 cycles) of 196 patients in the control group completed at least 3–6 cycles of treatment (Figure 1). Of these, 300 patients (n = 150 in each group) received the allocated treatment as adjuvant chemotherapy and 91 patients (experimental group, n = 45; control group, n = 46) received the allocated treatment as neoadjuvant chemotherapy (Supplemental Appendix 2, Table S1). The reasons for the discontinuation treatment among the 90 patients (experimental group, n = 53; control group, n = 37) were predominantly due to patient decision (21 vs 18), AEs (9 vs 7), investigator decision (9 vs 1; 6 in 9 due to a slower rate of CA-125 decline), 20 and allergic to study drug (8 vs 8). After completing study treatment, 44 patients (experimental group, n = 13; control group, n = 31) received poly (ADP-ribose) polymerase inhibitors (PARPi) as maintenance therapy (Supplemental Appendix 2, Table S2).
Efficacy
Primary efficacy endpoint
In the PPS population, the median PFS was 35.3 months (95% CI, 23.7–46.9) in the experimental group and 35.0 months (95% CI, 26.9–43.1) in the control group (HR = 0.99, 95% CI, 0.73–1.35; p = 0.94; Figure 2(a)). These results were consistent with the primary analysis PFS results in the FAS population, with an HR of 1.04 (95% CI, 0.79–1.38; Supplemental Appendix 2, Figure S1(A)).

Kaplan–Meier curves in the PPS population. (a) Kaplan–Meier curve of PFS. (b) Kaplan–Meier curves of OS.
Subgroup analysis of PFS according to stratification factors and patients’ characteristics was generally consistent with the primary findings (Figure 3(a) and Supplemental Appendix 2, Figure S2). Of note, 95% CIs for the HRs included 1.00 in all subgroups defined by stratification factors and almost all subgroups defined by demographic and baseline characteristics (Figure 3(a) and Supplemental Appendix 2, Figure S2). Additionally, univariate and multivariate analyses showed that FIGO stage, baseline residual disease, and breast cancer gene (BRCA) mutation status based on germline analysis were independent prognostic factors affecting PFS, irrespective of PPS or FAS population (Supplemental Appendix 2, Tables S3 and S4).
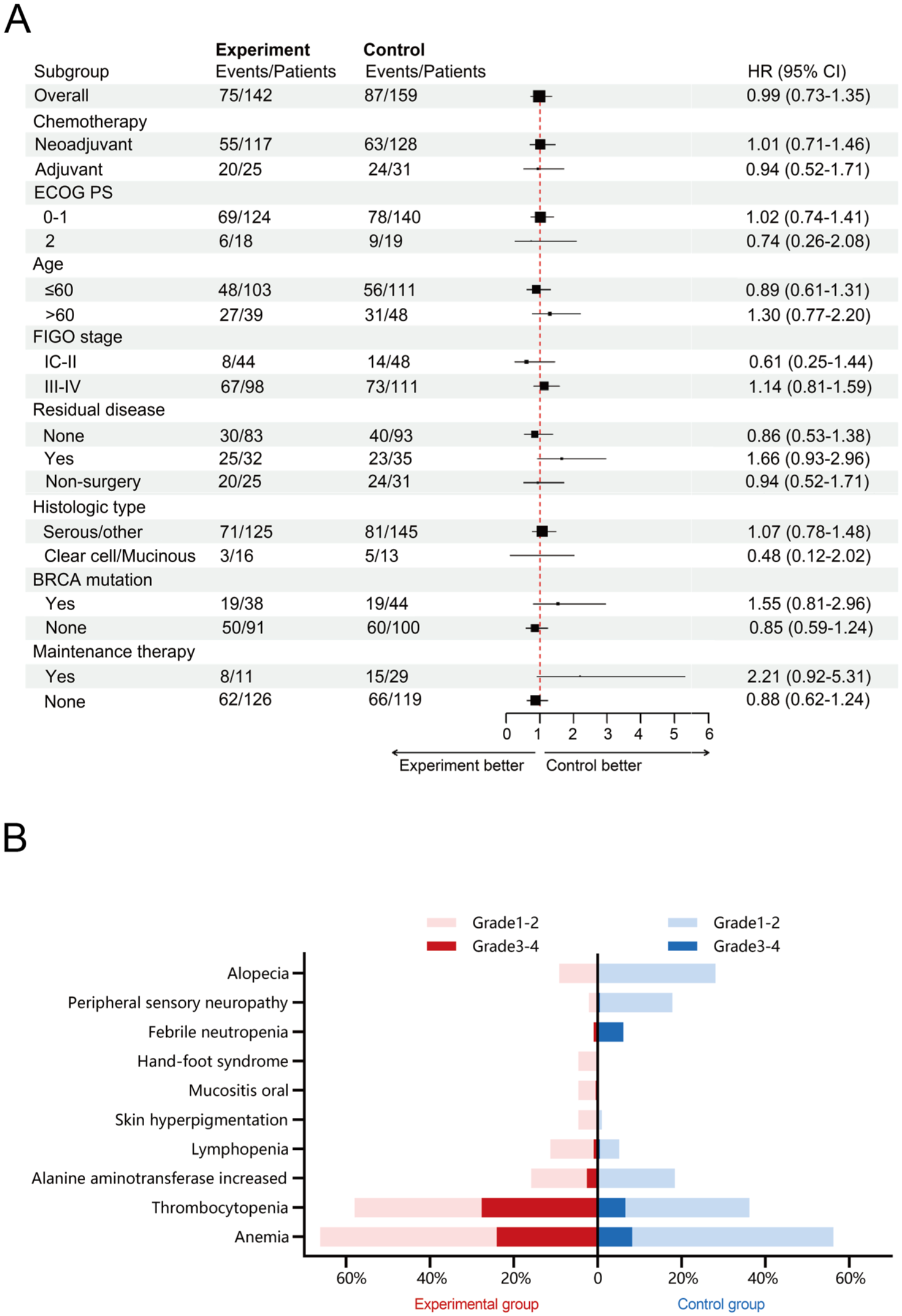
(a) Subgroup analyses of PFS in the PPS population. The unknown subgroup isn’t presented because of insufficient events. (b) Statistically significant AEs. PLD plus carboplatin significantly improved quality of life regarding alopecia and neurotoxicity in epithelial ovarian cancer patients, but demonstrated stronger hematologic toxicity compared to the control group.
Secondary efficacy endpoints
Tumor response as per RECIST version 1.1 was observed in patients with measurable lesions (Table 2). In the PPS population, the ORRs for patients with at least one target lesion in the experimental group and control group were 80.5% and 79.2%, respectively (p = 0.88). The DCRs were 90.2% and 83.3%, respectively (p = 0.34). For patients with only nontarget lesions only, the CR rates in the experimental group and control group were 61.3% and 60.0%, respectively (p = 0.92).
Efficacy outcomes as per response evaluation criteria in solid tumors version 1.1 in per protocol set.

There were four patients in the experimental group and three patients in the control group who had no radiographic examinations at baseline. Data are n (%).
CR, complete response; DCR, disease control rate; NE, not evaluate; ORR, objective response rate; PD, progressive disease; PR, partial response; SD, stable disease.
The activity was also observed in patients with elevated CA-125 levels as per GCIG criteria in the PPS population. For patients with baseline CA-125 levels ⩾2× upper limit of normal, the response rate was 96.6% in the experimental group and 100.0% in the control group, with no statistically significant difference between the two groups (p = 0.19; Supplemental Appendix 2, Table S6). Among the 193 patients with abnormal baseline CA-125 levels who returned to normal during the treatment period, the median time to normalization was 2 cycles in both groups.
At the time of analysis (July 30, 2024), 31 (experimental group, n = 11; control group, n = 20) deaths had occurred in the PPS population. The median OS was not reached in either group (HR = 0.60, 95% CI, 0.29–1.25; p = 0.16; Figure 2(b)). The 2-year and 4-year OS rates in the experimental group were 96.2% (95% CI, 91.0–98.4) and 87.6% (95% CI, 77.1–93.5), respectively, compared to 92.4% (95% CI, 86.7–95.7) and 82.4% (95% CI, 73.4–88.7) in the control group (all p > 0.05). All secondary efficacy endpoints in the FAS population (Supplemental Appendix 2, Tables S5 and S6 and Figure S1(B)) were consistent with those in the PPS population.
Safety
The overall safety profiles were similar between the two treatment groups (Table 3), with 84.6% (165/195) of patients in the experimental group and 86.2% (169/196) of patients in the control group reporting at least one AE. The most frequently observed AEs of any grade in the experimental and control groups were anemia (66.2% vs 56.1%), leukopenia (64.1% vs 61.7%), and neutropenia (60.0% vs 57.7%). We presented statistically significant AEs in Figure 3(b). It is worth mentioning that alopecia (9.2% vs 28.1%, p < 0.001), peripheral sensory neuropathy (2.1% vs 17.9%, p < 0.001), and febrile neutropenia (1.0% vs 6.1%, p = 0.01) of any grade reported in the experimental group was significantly lower than in the control group. Mucositis oral (4.6% vs 0.0%, p = 0.01), skin hyperpigmentation (4.6% vs 1.0%, p = 0.03), lymphopenia (11.3% vs 5.1%, p = 0.03), and hand-foot syndrome (4.6% vs 0%, p = 0.002) of any grade occurred more in the experimental group, with the most cases being of grades 1–2 severity. Grades 3–4 AEs were reported in 108 (55.4%) in the experimental group and 101 (51.5%) patients in the control group, respectively, with neutropenia (30.3% vs 35.2%) and thrombocytopenia (27.7% vs 6.6%) being the most common. No treatment-related death developed in either group. Dose modifications and dose delays owing to AEs occurred in 40 (20.5%) and 55 (28.2%) patients in the experimental group and 29 (14.8) and 44 (22.4%) patients in the control group, respectively. A similar proportion of patients in the experimental (4.6%) and control (3.6%) groups experienced treatment discontinuation due to AEs. The incidence of hypersensitivity to study drugs was also similar between the two groups, with 5.1% (10/195) for PLD and 5.1% (10/196) for paclitaxel.
Adverse events.

Data are shown for AEs that occurred in >1% of the patients in either group (except where noted) during the trial intervention or up to 30 days after discontinuation of the intervention. The events are listed in descending order of frequency in the experimental group.
p, grade 3–4.
p, all grade.
AE, adverse event.
Discussion
To the best of our knowledge, this trial was the first head-to-head comparison of PLD plus carboplatin with paclitaxel plus carboplatin as the first-line treatment for epithelial ovarian cancer in China. In this trial, the primary endpoint PFS was comparable between the PLD plus carboplatin and the paclitaxel plus carboplatin regimen. Additionally, there were no discernible differences in ORR, DCR, or OS between the two groups. Equally important was the lower incidence of long-term toxicities associated with PLD plus carboplatin, including alopecia and peripheral sensory neuropathy. The comparable efficacy and better safety profile of PLD plus carboplatin suggest it was a viable alternative, balancing effectiveness with tolerability. This trial offers valuable insights for personalized epithelial ovarian cancer treatment in China.
The median PFS was 35.3 months (95% CI, 23.7–46.9) in experimental group and 35.0 months (95% CI, 26.9–43.1) in the control group, respectively; the between-group difference met the predefined margin for noninferiority of PLD plus carboplatin treatment versus paclitaxel plus carboplatin treatment (HR = 0.99, 95% CI, 0.73–1.35; p = 0.94). The FAS encompasses all randomized subjects, adhering to the intention-to-treat principle to preserve randomization integrity and ensure result generalizability, primarily assessing drug efficacy and safety. Conversely, the PPS includes only subjects who strictly comply with the trial protocol, reflecting treatment effects under ideal conditions and serving as a sensitivity analysis to validate FAS robustness. This study aims to elucidate the drug’s biological impact and therapeutic efficacy, thus presenting PPS data to evaluate treatment outcomes. While this approach may introduce selection bias, utilizing FAS risks underestimating efficacy. Combining FAS and PPS ensures a comprehensive assessment of drug performance. This consistency was observed across both the PPS and FAS analyses, underscoring the robustness of the results irrespective of protocol adherence. Subgroup analyses of PFS also yielded similar results to the total study population, with 95% CIs for the HRs including 1.00 in all subgroups defined by stratification factors and almost all subgroups defined by demographic and baseline characteristics. These findings indicated that the therapeutic effects of both regimens are comparable across a broad spectrum of patient profiles.
The MITO-2 trial was the first study to study the substitution of paclitaxel with PLD (Caelyx®) in combination with carboplatin, adding important evidence about the role of PLD in the first-line treatment of epithelial ovarian cancer. While we acknowledge the limitations of cross-trial comparisons, the median PFS of 35.3 months in the experimental group was notable for being superior to the PFS (19.0 months) of PLD plus carboplatin in the MITO-2 trial. 14 One plausible reason for the higher median PFS observed in our study compared to the MITO-2 trial could be the higher proportion of early-stage (FIGO Stage IC–II) patients enrolled in our cohort (100/391 vs 153/820 in MITO-2). Similarly, for patients with clinically measurable disease, the ORR (80.5% vs 68%) of the experimental group in our trial was also superior to that of the PLD plus carboplatin in the MITO-2 trial. 21 Several factors may account for this difference, such as a higher proportion of patients had stage IC disease (13.8% vs 9.0%) and a higher no residual disease rate (53.3% vs 36.6%) in our study compared to those in the MITO-2 trial, etc. The higher no residual disease rate likely played a key role in mitigating surgical confounders, allowing for a more accurate evaluation of drug efficacy which eliminated the potential impact of surgery-related complications and residual tumors on patient prognosis. Another possible explanation might be our use of Duomeisu®, a domestically developed PLD from China. Despite its bioequivalence to Caelyx®, 15 any differences in clinical efficacy remain unclear and require further investigation through head-to-head clinical trials. Additionally, differences in patient populations could contribute to variations in treatment efficacy, as our study was conducted in a Chinese population, while the MITO study involved patients from other countries. A major strength of our study was the representativeness of the cohort, particularly in disease staging and pathological subtypes, which closely aligns with real-world epithelial ovarian cancer treatment and enhances the clinical relevance and practical applicability of our findings.
Our trial also confirmed that FIGO stage, age, baseline surgical situation, and BRCA mutation status based on germline analysis were independent prognostic factors in epithelial ovarian cancer, aligning with findings from previous studies.22–24 The BRCA1/2 mutation rate in the Chinese ovarian cancer population has been reported to be 28.5%. 25 The study confirmed a balanced distribution of this biomarker between the experimental and control groups at baseline (Table 1). Furthermore, no significant difference in PFS was observed between the experimental and control groups, irrespective of BRCA mutation status, suggesting that BRCA mutation status did not affect the drug selection, and indicating that PLD plus carboplatin was also equally effective in both BRCA-mutated and non-BRCA-mutated patients. These findings underscore the need for further research to elucidate optimal treatment strategies that can tailor therapy to individual patient profiles, enhancing both efficacy and patient quality of life.
The proportion of patients who received PARPi as maintenance therapy was notably low in both groups (6.7% vs 15.8%), which may be attributed to the limited clinical adoption of PARPi for maintenance therapy in ovarian cancer when the study was initiated. While this low utilization rate could potentially introduce confounding effects on the interpretation of PFS outcomes, our actual study data demonstrated no significant difference in PFS between the experimental and control groups with regard to PARP inhibitor application (Supplemental Appendix 2, Figure S2). Nonetheless, with disease recurrence and the initiation of subsequent therapies, including PARPi, there may be a potential impact on OS. Future analyses should account for these factors, as treatment decisions in later stages may introduce confounding effects on long-term survival outcomes.
Both treatment groups were generally well tolerated in our patients with epithelial ovarian cancer, with only a few patients (4.6% vs 3.6%) discontinuing treatment due to AEs. It may be related to our effective pretreatment protocol, such as prophylactic corticosteroid administration to both experimental and control groups of ovarian cancer patients to prevent hypersensitivity reactions. The safety profile in both treatment groups was consistent with the known side effects in previous studies. As expected, the experimental group experienced higher rates of mucositis oral (4.6% vs 0.0%) and leukopenia (45.1% vs 41.8%), consistent with previous studies of PLD.26,27 However, these AEs were generally short-term and manageable. In contrast, the control group showed a higher incidence of potentially long-term toxicities, including alopecia (9.2% vs 28.1%, p < 0.001) and peripheral sensory neuropathy (2.1% vs 17.9%, p < 0.001), which was consistent with that reported in the previous studies.12,28,29 These long-term toxicities might significantly impact on patient’s quality of life. Alopecia was notably one of the most distressing side effects of chemotherapy for many women. 30 Additionally, peripheral neuropathies were very common in the population ⩾65 years old, and their prevalence increases with age, particularly in patients with malignancies.31,32 Interestingly, in our study, a higher incidence of peripheral sensory neuropathy (17.9%) was observed with paclitaxel plus carboplatin compared to PLD plus carboplatin (2.1%, p < 0.001), suggesting that PLD plus carboplatin could offer a more advantageous treatment option for patients aged ⩾65 years. Importantly, no treatment-related deaths and new safety signals were observed in either group. Taken together, the safety profile of PLD plus carboplatin suggests a potential for improved quality of life in patients with epithelial ovarian cancer, especially regarding alopecia and neuropathy toxicity.
Our findings establish carboplatin-PLD as an effective and better-tolerated, noninferior alternative to carboplatin-paclitaxel for the first-line treatment of patients with epithelial ovarian cancer who are unsuitable for paclitaxel due to preexisting conditions such as neuropathy. It is crucial to emphasize that providing this alternative for this specific population does not deprive them of standard care but represents a necessary strategy when the standard regimen is contraindicated. It is important to recognize that the efficacy of maintenance therapies, particularly PARP inhibitors, is primarily determined by platinum sensitivity and Homologous Recombination Deficiency (HRD) status, rather than the specific partner drug used in the platinum-based doublet. Consequently, patients who respond to first-line carboplatin-PLD remain eligible for subsequent maintenance strategies without compromise.
This study had several limitations. First, all patients in our study were Chinese, potentially limiting the generalizability of our results to other racial/ethnic groups. Second, the OS data were immature at the cut-off date, further follow-up is needed to report a more mature assessment of OS. A final consideration is the incomplete BRCA testing rates in both groups (experimental group, n = 1; control group, n = 6), which poses a potential source of unrecognized bias. As BRCA mutation status is a known prognostic factor and can correlate with platinum sensitivity, 33 its absence could theoretically confound outcomes. To address this concern, we conducted a comparative analysis of baseline characteristics between patients with and without available BRCA data. The absence of significant differences between these subgroups (Table 1) suggests that the missing data are likely random rather than systematic. Nevertheless, this limitation should be considered when interpreting the study results.
Conclusion
Given the superior tolerability of the PLD plus carboplatin over paclitaxel plus carboplatin and their comparable efficacy as first-line treatment for epithelial ovarian cancer, substituting paclitaxel with PLD (Duomeisu®) in combination with carboplatin is both feasible and potentially more beneficial. This regimen could represent a novel treatment option for patients with epithelial ovarian cancer in China, and its manageable safety profile further supports its widespread clinical application.
Supplemental Material
sj-docx-1-tam-10.1177_17588359261420402 – Supplemental material for Pegylated liposomal doxorubicin plus carboplatin versus paclitaxel plus carboplatin as first-line therapy for epithelial ovarian cancer (CGCS-04): a multicenter, randomized clinical trial
Supplemental material, sj-docx-1-tam-10.1177_17588359261420402 for Pegylated liposomal doxorubicin plus carboplatin versus paclitaxel plus carboplatin as first-line therapy for epithelial ovarian cancer (CGCS-04): a multicenter, randomized clinical trial by Yuanming Shen, Nalan Tu, Beihua Kong, Bairong Xia, Yonghui Zou, Rutie Yin, Qingshui Li, Ying Yue, Shan Kang, Ke Wang, Liping He, Shixuan Wang, Yuanguang Meng, Pengpeng Qu, Xianghua Huang, Ruixia Guo, Ping Yan, Mei Pan, Wenjun Cheng, Ge Lou and Zehua Wang in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359261420402 – Supplemental material for Pegylated liposomal doxorubicin plus carboplatin versus paclitaxel plus carboplatin as first-line therapy for epithelial ovarian cancer (CGCS-04): a multicenter, randomized clinical trial
Supplemental material, sj-docx-2-tam-10.1177_17588359261420402 for Pegylated liposomal doxorubicin plus carboplatin versus paclitaxel plus carboplatin as first-line therapy for epithelial ovarian cancer (CGCS-04): a multicenter, randomized clinical trial by Yuanming Shen, Nalan Tu, Beihua Kong, Bairong Xia, Yonghui Zou, Rutie Yin, Qingshui Li, Ying Yue, Shan Kang, Ke Wang, Liping He, Shixuan Wang, Yuanguang Meng, Pengpeng Qu, Xianghua Huang, Ruixia Guo, Ping Yan, Mei Pan, Wenjun Cheng, Ge Lou and Zehua Wang in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-3-tam-10.1177_17588359261420402 – Supplemental material for Pegylated liposomal doxorubicin plus carboplatin versus paclitaxel plus carboplatin as first-line therapy for epithelial ovarian cancer (CGCS-04): a multicenter, randomized clinical trial
Supplemental material, sj-docx-3-tam-10.1177_17588359261420402 for Pegylated liposomal doxorubicin plus carboplatin versus paclitaxel plus carboplatin as first-line therapy for epithelial ovarian cancer (CGCS-04): a multicenter, randomized clinical trial by Yuanming Shen, Nalan Tu, Beihua Kong, Bairong Xia, Yonghui Zou, Rutie Yin, Qingshui Li, Ying Yue, Shan Kang, Ke Wang, Liping He, Shixuan Wang, Yuanguang Meng, Pengpeng Qu, Xianghua Huang, Ruixia Guo, Ping Yan, Mei Pan, Wenjun Cheng, Ge Lou and Zehua Wang in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We thank all clinical and pathology teams at participating sites of the NCT03794778, as well as the women who participated in the trials and their families. We thank for the technical support of CSPC Ouyi Pharmaceutical Co., Ltd.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.