
Abstract
Background:
Regorafenib targets tumor angiogenesis and oncogenic signaling by inhibiting multiple kinases involved in angiogenesis, oncogenesis, and the tumor microenvironment. Chemotherapy of FOLFIRI causes direct cytotoxic damage to tumor cells.
Objectives:
We evaluated the efficacy and safety of regorafenib plus FOLFIRI with uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) genotyping-guided irinotecan dose escalation in patients with metastatic colorectal cancer (mCRC) who had received second- or third-line systemic therapy.
Design:
A total of 153 patients were randomized (at a ratio of 2:1) to receive either regorafenib plus FOLFIRI (experimental group, 102 patients) or regorafenib alone (control group, 51 patients).
Methods:
Both groups received regorafenib (120 mg daily) for 21 consecutive days in 28-day cycles. In addition, the experimental group received FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation. The primary outcome was progression-free survival (PFS). The secondary outcomes were overall survival (OS), disease control rate (DCR), and adverse events.
Results:
The final cohort included 116 patients. The experimental group exhibited significant improvement in PFS (p = 0.016) but marginally significant improvements in DCR (p = 0.055). Specifically, PFS improved significantly for experimental group patients carrying wild-type rat sarcoma virus (RAS; p = 0.003) and those carrying left-sided colon tumors (p = 0.015). A trend toward improved OS was noted in experimental group patients carrying wild-type RAS (p = 0.096). No significant between-group difference was observed in the incidence of severe adverse events (all p > 0.05).
Conclusion:
Our findings suggest that regorafenib plus FOLFIRI significantly improved PFS in patients with mCRC and offers a safety profile similar to that of regorafenib alone. This regimen may be particularly beneficial for patients with mCRC carrying wild-type RAS and those carrying left-sided tumors. However, larger phase III randomized controlled trials are necessary to confirm the efficacy and safety of this combination therapy before adoption into standard practice.
Trial registration:
ClinicalTrials.gov registry: NCT03880877 (https://clinicaltrials.gov/search?cond=NCT03880877).
Plain language summary
This study explored a new treatment strategy for metastatic colorectal cancer (mCRC) in patients whose disease progressed after prior therapies. We investigated combining regorafenib, a drug that targets cancer growth and blood vessel development, with FOLFIRI chemotherapy, which directly kills cancer cells. The FOLFIRI regimen was tailored using a UGT1A1 genetic test to personalize irinotecan dosing. In this trial, 153 patients were randomly assigned to receive either regorafenib plus FOLFIRI or regorafenib alone. Our findings show that the regorafenib plus FOLFIRI group had significantly longer progression-free survival (PFS), meaning their cancer was controlled for a longer period. This benefit was even more pronounced in patients with wild-type RAS tumors and those with left-sided colon tumors. Importantly, the combination therapy did not significantly increase severe side effects compared to regorafenib alone. These results suggest that combining regorafenib with FOLFIRI could offer a valuable new option for mCRC patients, particularly those with wild-type RAS and left-sided tumors. However, larger studies are needed to confirm these findings and establish this combination as a standard treatment.
Introduction
Most patients with metastatic colorectal cancer (mCRC) require systemic therapy. Current systemic chemotherapy for mCRC includes FOLFOX and FOLFIRI regimens, which combine 5-fluorouracil/leucovorin with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI). These regimens have been associated with a median overall survival (OS) of approximately 20 months.1 –3 The addition of biological agents, such as vascular endothelial growth factor (VEGF) inhibitors or epidermal growth factor receptor (EGFR) inhibitors, to FOLFOX or FOLFIRI (administered as standard first- or second-line therapy) has been demonstrated to improve oncological outcomes compared with those achievable with chemotherapy alone, with a reported median OS of approximately 30 months.4,5 Immunotherapy is remarkably effective in patients with CRC whose tumors exhibit defective mismatch repair or high microsatellite instability. 6
Regorafenib, a small-molecule multikinase inhibitor, is an oral drug that impedes tumor angiogenesis, proliferation, growth, and metastasis.7,8 In 2012, the US Food and Drug Administration (FDA) approved the use of regorafenib as a third- or later-line therapeutic agent for patients with mCRC who were refractory to FOLFOX, FOLFIRI, or monoclonal antibodies against VEGF or EGFR. 9 In 2013, Taiwan’s FDA approved the use of regorafenib for this purpose. 10 Phase III clinical trials have demonstrated that regorafenib monotherapy substantially improved the disease control rate (DCR), progression-free survival (PFS), and OS in patients with refractory mCRC.11,12 In Taiwanese patients with mCRC receiving regorafenib monotherapy, the reported median OS and PFS were 11.64 and 2.17 months, respectively. 13
Uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) genotyping serves as a predictive marker of severe irinotecan-related adverse effects (AEs), such as diarrhea and neutropenia. 14 We previously reported that UGT1A1 genotyping-guided irinotecan dose escalation improves both PFS and OS and that this dose adjustment is an independent factor influencing DCR.15,16 Based on these findings, we began treating patients with refractory mCRC with regorafenib plus FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation.17,18 Our retrospective study revealed that this regimen extended PFS and OS compared with those reported by the CORRECT 11 and CONCUR trials. 12
The rationale for combining FOLFIRI (a cytotoxic chemotherapy) with regorafenib (a multi-kinase inhibitor) is grounded in the goal of enhancing efficacy through dual mechanisms. Regorafenib targets tumor angiogenesis and oncogenic signaling by inhibiting multiple kinases involved in angiogenesis, oncogenesis, and the tumor microenvironment. These are not directly targeted by standard chemotherapy. However, FOLFIRI causes direct cytotoxic damage to tumor cells. Irinotecan inhibits topoisomerase I, leading to DNA damage in rapidly dividing cells, while 5-FU and leucovorin disrupt RNA function and DNA synthesis. Combining these two offers a synergistic approach because regorafenib may normalize tumor vasculature, improving chemotherapy delivery, and may also modulate the tumor microenvironment, potentially enhancing sensitivity to chemotherapy. On the basis of our previous studies, we hypothesized that UGT1A1 genotyping-guided irinotecan dose escalation would improve clinical outcomes without significantly increasing toxicity in patients receiving regorafenib plus FOLFIRI for refractory mCRC. Therefore, in the present study, we compared the efficacy and safety of regorafenib plus FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation with regorafenib alone in patients with refractory mCRC who have already received second- or third-line systemic therapy. 19
Patients and methods
Study design
This multicenter, phase II, open-label, two-arm randomized controlled trial was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki and in adherence to local laws and regulations consistent with the International Conference on Harmonization Good Clinical Practice Guidelines and other regulatory requirements. The study protocol was approved by the institutional review boards of Kaohsiung Medical University Hospital (permit number: KMUHIRB-F(II)-20190032), China Medical University Hospital (permit number: CMUH108-REC2-124), and Tainan Municipal Hospital (permit number: 1100709). The trial was registered with ClinicalTrials.gov (identifier: NCT03880877).
The detailed inclusion and exclusion criteria, treatment regimens, and study outcomes have been previously reported in our protocol. 19 The escalation design was based on our previous studies.10,16 –18 Taiwan’s National Health Insurance guideline recommends regorafenib as a fourth-line therapy for patients with mCRC carrying wild-type rat sarcoma virus (RAS) and a third-line therapy for those carrying mutant RAS. Therefore, all patients in the two arms had previously received irinotecan. Patients with mCRC carrying wild-type RAS had received target therapy of both anti-EGFR agents and anti-VEGF agents. Patients with mCRC carrying mutant RAS had received target therapy of anti-VEGF agents. Figure 1 depicts the study design. The patients were randomized (at a ratio of 2:1) to receive either regorafenib plus FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation (experimental group) or regorafenib alone (control group; Figure 2). For patients with UGT1A1 *1/*1 and *1/*28, if no FOLFIRI-related AEs greater than grade 3 are observed after two cycles at a given irinotecan dose, the dose will be escalated in increments of 30 mg/m2. If any FOLFIRI-related AEs greater than grade 3 occur, the irinotecan dose will be reduced by 30 mg/m2 or returned to the previous dose and maintained at that level for the next cycle. Specifically, patients with UGT1A1 *1/*1 received irinotecan starting at 180 mg/m2, with escalation to 260 mg/m2 if tolerated. For patients with *1/*28, the starting dose was 150 mg/m2 with a maximum of 240 mg/m2. Those with *28/*28 received a reduced dose of 120 mg/m2 with no escalation. Both groups received regorafenib at a consistent dose of 120 mg for 21 consecutive days (days 1–21) of each 28-day cycle. 19

Treatment plan.

CONSORT diagram.
The sample size was estimated using the log-rank test for survival analysis, assuming exponential survival distributions in both groups. The primary endpoint was PFS. We hypothesized a median PFS of 3.2 months in the control group and 7.0 months in the experimental group, using a two-sided significance level (α) of 0.25 and 90% power (1–β = 0.90). These parameters were selected to reflect the exploratory nature of the study and our focus on identifying clinically meaningful differences. The overall event rate was estimated at 0.77, based on the assumption that 10 out of 13 enrolled patients would experience disease progression by the end of the study. Under these assumptions, the required sample size was calculated to be 121 patients. To account for a potential 20% dropout rate, the target sample size was increased to 153 patients, with a 2:1 randomization ratio (experimental group:control group = 102:51). Sample size calculations were performed using the powerSurvEpi package in R (version 4.3.1; R Foundation for Statistical Computing, Vienna, Austria), a validated tool for power analysis in time-to-event outcomes. The recruitment target could be adjusted on the basis of the actual rate of patient enrollment.
Study outcomes
The primary outcome was investigator-assessed PFS, whereas the secondary outcomes were OS, DCR, and AEs.
Treatment response assessment
Treatment responses were assessed in accordance with routine clinical practice, typically every 8 weeks. A radiologist categorized treatment responses using the Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1). 20 AEs were monitored and graded in each treatment cycle in accordance with the National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCT-CTCAE, version 5; https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm).
Study populations for data analysis
Efficacy outcomes were primarily analyzed using data from the per-protocol (PP) population. The study cohort was divided into intention-to-treat (ITT) and PP populations. In the ITT population, all patients were randomized. The PP population was a subset of the ITT population and included patients who had no major protocol violations and demonstrated drug compliance for at least two cycles.
Statistical analysis
All data were analyzed using SPSS (version 22.0; IBM Corporation, Armonk, NY, USA). Between-group comparisons of various clinicopathological characteristics were performed using the Chi-square test for categorical variables. Univariate and multivariate logistic regression models were used to identify independent predictors of disease progression and mortality. PFS was defined as the interval between treatment initiation and the first record of radiological progression or death, whichever occurred first. OS was defined as the interval between treatment initiation and death due to any cause. The DCR was defined as the proportion of patients whose best response indicated no disease progression (complete response, partial response, or stable disease). Stable disease was considered to be disease stability lasting for at least 6 weeks. Univariate and multivariate analyses for identifying independent prognostic factors for OS and PFS were performed using a Cox proportional hazards model. PFS and OS were evaluated using the Kaplan–Meier method. Between-group comparisons of time-to-event distributions were performed using a log-rank test. A p value of <0.05 was considered to be significant. The reporting of this study conforms to the SPIRIT-Outcomes 2022 Checklist (Table S1). 21
Results
Clinicopathological characteristics of the study groups
This study included 153 patients (experimental group: 98; control group: 55; Figure 2) from April 2019 to May 2023. In the experimental group, 20 patients either refused treatment or did not receive it due to personal reasons, and 4 patients received only one treatment session. In the control group, 12 patients either refused treatment or did not receive it due to personal reasons, and 5 patients received only one treatment session. As a result, only 116 patients were included in the per-protocol analysis. The clinicopathological characteristics and treatment responses of the PP population are summarized in Table 1. Finally, 116 patients were included in the analysis. Of them, 74 (63.8%) were in the experimental group, whereas 42 (32.6%) were in the control group. The baseline demographics and clinicopathological characteristics were similar between the two groups (all p > 0.05). Although dosing was determined prospectively based on genotyping, we retrospectively confirmed the ranges administered during the trial. This includes a starting dose of 180 mg/m2 for *1/*1 patients, escalating to 260 mg/m2, starting dose of 180 mg/m2 for *1/*28 patients, escalating to 210 mg/m2, and starting dose of 120 mg/m2 for *28/*28 patients, escalating to 150 mg/m2. The DCR was higher, although non-significant, in the experimental group than in the control group (55.4% vs 38.1%; p = 0.073). The proportion of patients who had received three lines of systemic therapy before receiving regorafenib was higher, although non-significant, in the experimental group than in the control group (44.6% vs 28.6%; p = 0.089).
Baseline clinicopathological characteristics and treatment responses of the treatment groups.

ECOG PS, Eastern Cooperative Oncology Group Performance Score; EGFR, epidermal growth factor receptor; mCRC, metastatic colorectal cancer; PD, progressive disease; PR, partial response; RAS, rat sarcoma virus; SD, stable disease; UGT1A1, uridine diphosphate glucuronosyl transferase 1A1.
Patient survival
Regorafenib plus FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation was identified to be an independent favorable prognostic factor for PFS (p = 0.008; hazard ratio (HR) = 1.892; 95% confidence interval (CI): 1.182–3.027; Table 2). According to intent-to-treat analysis, Kaplan–Meier survival analysis revealed that PFS was significantly longer in the experimental group than in the control group (4.7 vs 2.8 months; p = 0.003; Figure 3(a)). However, no significant difference in OS was noted between the two groups (7.1 vs 7.9 months; p = 0.966; Figure 3(b)).
Univariate and multivariable analysis of prognostic factors on PFS and OS.

Indicated p < 0.05.
CI, confidence interval; HR, hazard ratio; OS, overall survival; PFS, progression-free survival; RAS, rat sarcoma virus.

Kaplan–Meier survival curve for patients with refractory mCRC stratified by treatment group. According to intent-to-treat analysis (a) Progression-free survival (p = 0.003). (b) Overall survival (p = 0.966).
Moreover, according to PP analysis, Kaplan–Meier survival analysis revealed that PFS was significantly longer in the experimental group than in the control group (4.7 vs 2.8 months; p = 0.005; Figure 3(c)). However, no significant difference in OS was noted between the two groups (9.4 vs 10.7 months; p = 0.932; Figure 3(b)).
Subgroup analyses by RAS status were performed. For patients carrying wild-type RAS, PFS was significantly longer in the experimental group than in the control group (6.3 vs 2.1 months; p = 0.003; Figure 4(a)). However, for patients carrying mutant RAS, no significant between-group difference was observed in the PFS (Figure 4(c)). Furthermore, for patients carrying wild-type RAS (Figure 4(b)) and those carrying mutant RAS (Figure 4(d)), no significant between-group difference was observed in OS. A trend toward improved OS was observed in the experimental group patients carrying wild-type RAS (p = 0.096; Figure 4(b)).

Kaplan–Meier survival curve for patients with refractory mCRC stratified by treatment group and RAS gene status. (a) PFS of patients with mCRC showing RAS gene wild type stratified by treatment group (p = 0.003). (b) OS of patients with mCRC showing RAS gene wild type stratified by treatment group (p = 0.096). (c) PFS of patients with mCRC showing RAS gene mutation type stratified by treatment group (p = 0.136). (d) OS of patients with mCRC showing RAS gene mutation type stratified by treatment group (p = 0.124).
We further performed subgroup analyses by primary tumor location. For patients carrying left-sided colon tumors, PFS was significantly longer in the experimental group than in the control group (3.3 vs 2.7 months; p = 0.015; Figure 5(a)). However, for patients carrying right-sided colon tumors, no significant between-group difference was observed in the PFS (Figure 5(c)). Furthermore, for patients carrying left-sided colon tumors (Figure 5(b)) and those carrying right-sided colon tumors (Figure 5(d)), no significant between-group difference was observed in OS.

Kaplan–Meier survival curve for patients with refractory mCRC stratified by treatment group and primary tumor sidedness. (a) PFS of patients with left-sided mCRC stratified by treatment group (p = 0.015). (b) OS of patients with left-sided mCRC stratified by treatment group (p = 0.417). (c) PFS of patients with right-sided mCRC stratified by treatment group (p = 0.153). (d) OS of patients with right-sided mCRC stratified by treatment group (p = 0.056).
Safety of the combined regimen
The safety profiles for both groups are summarized in Table 3. The incidence of any-grade hematological AEs was significantly higher in the experimental group than in the control group (39.2% vs 0.00%; p < 0.001). However, the experimental and control groups were similar in terms of the incidence of any-grade non-hematological AE (59.5% vs 73.8%; p = 0.120). No significant between-group difference was observed in the incidence of severe hematological or non-hematological AEs (all p > 0.05).
Adverse events of patients according to treatment groups.
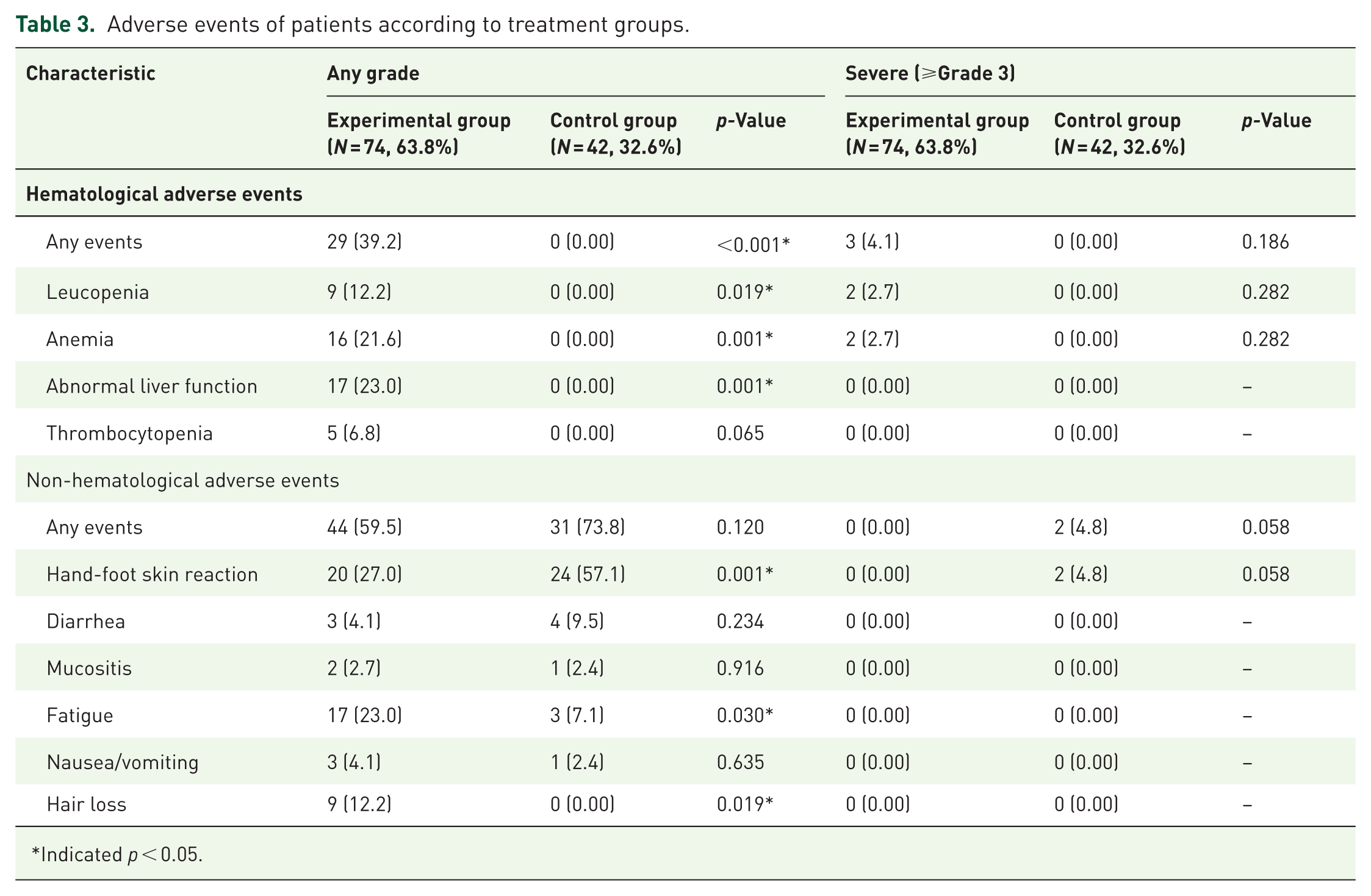
Indicated p < 0.05.
Discussion
To the best of our knowledge, this study is the first to evaluate the efficacy and safety of combining irinotecan with regorafenib, with irinotecan dosing tailored according to patients’ UGT1A1 genotypes. A significant improvement in PFS was observed in the combination therapy group. Subgroup analyses revealed that patients with wild-type RAS and those with left-sided primary tumors experienced notably prolonged PFS. Although the median OS did not differ significantly between the experimental and control groups, a trend toward improved OS was observed in wild-type RAS patients receiving the combination therapy. Furthermore, a marginally significant improvement in DCR was noted in the experimental group. Multivariate analysis identified regorafenib plus FOLFIRI as an independent predictor of disease control and an independent prognostic factor for PFS. Importantly, the addition of irinotecan did not result in a significant increase in the incidence of severe adverse events.
Over the past two decades, therapeutic advancements—including the use of metastasectomy—have extended the median OS for patients with mCRC from approximately 16 to 27.4–36 months.21,22 Previous studies have reported a median PFS of 1.7–3.9 months in patients receiving regorafenib monotherapy.11,12,23 –30 In the present study, the control group exhibited a median PFS of 2.8 months, which is consistent with these findings. Notably, the experimental group demonstrated a significantly longer median PFS of 4.7 months. Similarly, prior studies have reported a median OS of 5.0–9.3 months for patients treated with regorafenib monotherapy.11,12,23 –30 In our study, the control group had a median OS of 10.7 months, aligning with the upper range of previously reported outcomes. Regarding DCR, real-world data indicate a range of 24%–46% among patients with refractory mCRC receiving regorafenib monotherapy.23 –30 In our study, the control group achieved a DCR of 38.1%, consistent with the literature, while the study group achieved a notably higher DCR of 55.4%.
Since October 2013, we have administered regorafenib plus FOLFIRI, with UGT1A1 genotype-guided irinotecan dose escalation, as a treatment strategy for patients with refractory mCRC. 16 Given the high incidence of severe toxicities associated with the standard regorafenib dose of 160 mg/day-often necessitating dose reductions or treatment interruptions, as reported in the literature—we previously adjusted the dose to 120 mg/day. 17 This adjustment resulted in improved PFS and OS compared with outcomes reported in phase III clinical trials.11,12 Consequently, a regorafenib dose of 120 mg/day was adopted in the present study. Suzuki et al. 30 emphasized the importance of identifying an optimal regorafenib dose that balances tolerability and efficacy, avoiding treatment discontinuation due to toxicity. Supporting this, a phase I trial recommended a phase II dose of 120 mg/day for regorafenib. 30 At this dosage, the combination of regorafenib with chemotherapy was shown to be both feasible and safe as a second- to fourth-line treatment for mCRC, with reported median PFS ranging from 3.9 to 6.2 months.31 –34
We conducted subgroup analyses to identify patients most likely to benefit from the combination of regorafenib and FOLFIRI. Among patients with wild-type RAS, those in the experimental group experienced a significantly longer PFS and showed a non-significant trend toward improved OS compared to the control group (estimated median OS: 9.6 vs 6.6 months, p = 0.096). By contrast, for patients with RAS mutations, no significant differences in PFS or OS were observed between the two groups. Similarly, among patients with left-sided tumors, the experimental group demonstrated significantly prolonged PFS and comparable OS relative to the control group. However, for those with right-sided tumors, no significant differences were found in either PFS or OS between groups. These findings are consistent with those reported by Yoon et al., 35 who observed significantly longer PFS in patients with left-sided mCRC compared to those with right-sided tumors (2.6 vs 1.9 months).
A previous retrospective analysis by Ma et al., 18 which involved a smaller cohort of 41 patients, identified tumor sidedness as the sole independent predictor of clinical benefit from the combination therapy of FOLFIRI and regorafenib. However, this finding was not replicated in the present study. As shown in Table 3, tumor location (right- vs left-sided) did not reach statistical significance for OS in either univariate analysis (HR = 0.936, 95% CI: 0.548–1.598; p = 0.808) or multivariable analysis (HR = 0.873, 95% CI: 0.477–1.596; p = 0.873). Instead, two variables emerged as independent prognostic factors for OS: age ⩾65 years (HR = 1.659, 95% CI: 1.030–2.672; p = 0.037) and lung metastasis as the dominant metastatic site (HR = 0.501, 95% CI: 0.251–0.997; p = 0.049).
The discrepancy between the two studies may be attributable to differences in patient populations, sample sizes, statistical power, or methodological rigor. The limited sample size of the earlier study may have constrained the robustness of multivariate modeling, potentially overestimating the prognostic impact of tumor sidedness. By contrast, the larger cohort in the current study allowed for more comprehensive adjustment for covariates, providing a more nuanced evaluation of prognostic factors. The identification of age and lung metastasis as significant prognostic markers aligns with existing clinical evidence highlighting their relevance in shaping treatment outcomes for patients with mCRC. These findings underscore the importance of future prospective studies to further elucidate the prognostic and predictive roles of tumor sidedness and other clinical characteristics in the context of regorafenib-based combination therapy. Another study by Sanoff et al. 33 reported a second-line benefit of combining regorafenib with FOLFIRI compared to FOLFIRI alone in patients with wild-type RAS mCRC. This finding suggests that the benefit observed may not be solely due to the additive effects of the two agents but could potentially reflect a synergistic interaction. Regorafenib, a multikinase inhibitor targeting angiogenic, stromal, and oncogenic receptor tyrosine kinases, may modulate the tumor microenvironment and enhance the cytotoxicity of irinotecan-based chemotherapy. The current study does not explicitly test for synergy, but the authors’ findings—particularly the improved outcomes in specific subgroups such as older patients and those with lung metastases—raise the question of whether certain tumor or host characteristics could potentiate such an interaction. While the absence of significant predictive value for RAS status in this analysis tempers the generalizability of Sanoff’s findings, the possibility of synergistic benefit remains biologically plausible and warrants prospective evaluation with pharmacodynamic and biomarker correlatives.
This study has several limitations. First, a relatively high dropout rate reduced the initial sample size from 153 to 116 patients, thereby limiting the statistical power to detect significant differences between the study and control groups. Second, under Taiwan’s National Health Insurance guidelines, regorafenib is approved as a fourth-line therapy for patients with wild-type RAS and as a third-line therapy for those with mutant RAS. As a result, a larger proportion of patients in the study group received regorafenib as fourth-line therapy compared to the control group, potentially contributing to the absence of a significant difference in OS between the groups. In addition, the experimental group had a higher proportion of patients under 65 years of age and with wild-type RAS, factors that may have positively influenced OS outcomes. The imbalance in age and RAS status between arms may have introduced bias in the OS outcomes. Older age is generally associated with poorer prognosis and may have contributed to shorter OS in the affected group. Similarly, RAS mutations are known to be associated with more aggressive disease and reduced response to certain therapies, which could have negatively impacted survival outcomes in the arm with a higher proportion of RAS-mutant patients. In our cohort, a higher proportion of patients ⩾65 years were present in the control group (54.8%) compared to the experimental group (37.8%), although this difference did not reach statistical significance (p = 0.078). Multivariate Cox regression analysis confirmed that age ⩾65 years was an independent unfavorable prognostic factor for OS (HR = 1.659, 95% CI: 1.030–2.672, p = 0.037), suggesting that the older age distribution in the control arm may have contributed to the numerically lower median OS observed in that group. Similarly, RAS mutation status was imbalanced across arms, with more RAS-mutant patients in the control group (71.4%) than in the experimental group (55.4%; p = 0.089). Although RAS mutation status was not statistically significant in the multivariate analysis for OS (HR = 1.065, 95% CI: 0.657–1.728, p = 0.797), subgroup analyses revealed that patients with wild-type RAS derived greater benefit from the combination regimen in terms of PFS (6.3 vs 2.1 months; p = 0.003) and exhibited a trend toward improved OS (9.6 vs 6.6 months; p = 0.096). These findings suggest a differential treatment effect based on RAS genotype that may not be fully accounted for in the overall multivariate model. Therefore, although adjusted analyses were performed, the underlying imbalances in age and RAS status—both clinically relevant prognostic factors—likely had residual impact on survival outcomes. Third, data on the irinotecan dose in the experimental group were not collected. Fourth, data on subsequent treatments after disease progression were not collected, which may confound survival outcomes and obscure the full impact on OS. Fifth, the follow-up period was relatively short, with a median duration of 9.6 months and a mean duration of 12.5 months. Given the limited survival expectancy of patients with refractory mCRC undergoing later-line therapies, the study was only able to capture short-term (12 months) survival outcomes. Finally, we did not assess enzymatic activity either in vivo or in vitro, nor did we measure levels of irinotecan metabolites. Despite these limitations, the study offers meaningful insights—particularly from subgroup analyses—that help identify patient populations most likely to benefit from the combination of regorafenib and FOLFIRI. These findings may inform future research and clinical decision-making in the management of refractory mCRC.
Conclusion
To the best of our knowledge, this study is the first to evaluate the efficacy and safety of regorafenib plus FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation. Patients receiving this combination therapy demonstrated significant improvements in PFS and marginally significant improvements in DCR. Subgroup analyses suggested a potential trend toward improved outcomes with this regimen in patients with mCRC harboring wild-type RAS and those with left-sided tumors in the later-line setting; however, these findings did not reach statistical significance and should be interpreted with caution. Importantly, the addition of irinotecan did not result in a significant increase in severe adverse events, supporting the favorable safety profile of this personalized combination approach. However, larger phase III randomized controlled trials are necessary to confirm the efficacy and safety of this combination therapy before adoption into standard practice.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251371489 – Supplemental material for Efficacy and safety of regorafenib plus FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation against metastatic colorectal cancer: a multicenter, phase II, open-label, two-arm randomized controlled tria
Supplemental material, sj-docx-1-tam-10.1177_17588359251371489 for Efficacy and safety of regorafenib plus FOLFIRI with UGT1A1 genotyping-guided irinotecan dose escalation against metastatic colorectal cancer: a multicenter, phase II, open-label, two-arm randomized controlled tria by Ching-Wen Huang, Yen-Cheng Chen, Tao-Wei Ker, Yi-Wen Lin, Tsung-Kun Chang, Wei-Chih Su, Po-Jung Chen, Hsiang-Lin Tsai and Jaw-Yuan Wang in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.