
Abstract
Background:
Genome and transcriptome analysis has enhanced the characterisation of pancreatic ductal adenocarcinoma (PDAC), paving the way for targeted therapies. Tumours KRAS wild type (WT) represent a unique subgroup.
Objectives:
Characterise the population and molecular abnormalities present in KRAS WT PDAC.
Design:
Clinical and molecular data from a large retrospective cohort of KRAS WT PDAC were analysed.
Methods:
Next-generation sequencing (NGS) was used to analyse DNAs and RNAs, allowing molecular and transcriptomic characterisation.
Results:
We identified 93/1059 (9%) KRAS WT PDAC, among which eight had druggable fusions (n = 8/30 contributive samples), six had BRAF mutations and 19 (n = 19/47) had mutations in homologous recombination (HR) pathway genes. Potential molecular targets in this series may be underestimated due to many non-contributive results. Clinical characteristics and survival did not differ between patients with KRAS WT and KRAS-mutated tumours. Transcriptomic data were available for 350 samples. Their analysis shows a difference in phenotype between mutated and WT tumours, with a molecular profile that appears to be better prognostic for KRAS WT tumours.
Conclusion:
KRAS WT tumours are enriched with molecular abnormalities of therapeutic interest. These include oncogene driver alterations (gene fusions and mutations) and mutations in genes of the HR pathway. Targeted therapy strategies for PDAC rely on molecular testing beyond RAS, but further research is needed to identify new therapeutic approaches that improve outcomes in PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for 90% of pancreatic cancers and nearly 3% of cancers worldwide. 1 The increase in its incidence and its poor prognosis make this disease a public health problem. 2 The only curative treatment is surgery, but most patients are diagnosed at an advanced stage of the disease, and only systemic treatments can be offered. 3 Therapeutic progress has been made in recent years thanks to changes in chemotherapy protocols and the development of therapies targeting somatic or germline molecular abnormalities.4–6 Examples include zenocutuzumab for NRG1 fusion 7 or entrectinib for NTRK fusion 8 but also PARP inhibitors validated in patients with a deleterious germline BRCA1/2 mutation 9 or pembrolizumab in patients with deficient MisMatch Repair (dMMR)/MicroSatellite Instability (MSI) PDAC. 10 Pishvaian et al. 11 demonstrated that treatments adapted to molecular profiles resulted in a survival benefit. However, these tumour subgroups represent only a small proportion of PDAC.12–14
KRAS activating mutations have been tightly linked to PDAC carcinogenesis, are present in nearly 90% 15 for samples and are known to be an early molecular event. It is associated with other somatic mutations, such as SMAD4, TP53 and CDKN2A, which cannot be targeted by specific treatments. 16 The KRAS G12C mutation is currently accessible to targeted therapy but found in less than 5% of PDAC, and early development is ongoing with RAS multi-inhibitor.17,18 Conversely, KRAS wild-type (WT) tumours, representing about 10% of PDAC, are enriched in targetable molecular alterations. These include point mutations, gene fusions and gene amplifications.19–21 The management of this sub-population of PDAC could therefore be improved by adapting treatments according to tumour genotypes, as is the case for other cancers such as colorectal cancer or lung cancer. However, there are only a few cohorts of KRAS WT PDAC in the literature.22–26 Better characterisation, both genomic and transcriptomic, of these tumours is therefore necessary to be able to hope for an evolution of management in these patients. Integrative DNA/RNA analysis may help classify PDAC to identify patients who are likely to benefit from targeted therapies.
This work aimed to assess the diversity of molecular alterations in KRAS WT to ultimately guide treatments in patients with PDAC.
Materials and methods
Population
We retrospectively used data on patients and their tumours from pre-existing cohorts that had already been studied or were currently being studied. Patients came from different French and Belgian centres and were included in the studies between 1996 and 2016. We selected both patients with resected PDAC and patients with metastatic disease. To include patients in our analysis, sufficient tumour material was required to determine KRAS status. The following cohorts were used:
- For resected PDAC: the prospective multicentric cohort of Puleo et al. 27 led to the definition of a molecular classification of PDAC; the prospective multicentric cohort of Zhao et al. 28 was used to assess the prognostic relevance of the transcriptomic components previously described by Puleo et al.; and the prospective multicentric PRODIGE24 study sponsored by Unicancer and CCTG and published by Conroy et al. 6 compared modified FOLFIRINOX to gemcitabine in adjuvant. Genomic and transcriptomic data for PRODIGE24 patients were available through the MOSAPAC research programme. 29
- For metastatic disease: a prospective single-centre cohort of metastatic PDAC from the Pitié Salpêtrière hospital group 30 ; patients from the prospective multicentric phase II trials PRODIGE 35 (PANOPTIMOX) 31 and PRODIGE 37 (FIRGEMAX) 32 compared several chemotherapy regimens in the metastatic first line.
Collection of clinical data
The demographic and pathological data available for all included patients were compiled to identify clinical or histological features in patients with KRAS WT PDAC. Data of interest for patients with resected tumours included the following: age at disease onset, gender, tumour size, tumour differentiation, TNM stage, resection margin, adjuvant therapy received, recurrence-free survival (RFS) and overall survival (OS). For patients with metastatic disease, we analysed the following: age, gender, WHO status, biological parameters (neutrophil count (/mm3), haemoglobin (g/dL), albumin (g/L), total bilirubin (μmol/L), CA19.9 (U/L) and CEA (U/L)), treatment received and OS.
Determination of KRAS status and other mutations
KRAS status was determined by next-generation DNA sequencing (NGS) or quantitative polymerase chain reaction (qPCR) (Idylla Platform, Biocartis®, Belgium). DNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tumour blocks using the QIAamp® DNA FFPE Tissue Kit (QIAGEN®, Germany). All tumours that were either non-contributive or WT by Idylla were sequenced. Tumours whose KRAS results could not be interpreted by qPCR or KRAS WT tumours identified by qPCR were then sequenced by NGS to confirm the absence of a KRAS mutation. In addition, from the sequencing data, we were able to identify other pathogenic mutations present in KRAS WT tumours. We used different Ampliseq targeted panels and Ion Torrent™ sequencing (Thermo Fisher Scientific®, USA) to analyse mutational profiles. The list of panels is available as Supplemental Information (Table S1). All variants were detected with >99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of >300× and an analytic sensitivity of 5%. Genetic variants identified were categorised as ‘pathogenic’, ‘likely pathogenic’, ‘variant of unknown significance’, ‘likely benign’ or ‘benign’, according to the American College of Medical Genetics and Genomics standards. ‘Pathogenic’ and ‘likely pathogenic’ were reported as mutations, while ‘benign’, ‘likely benign’ variants and ‘variants of unknown significance’ were excluded.
Identification of gene fusions
We assessed gene fusions in KRAS WT tumours. We extracted RNA from formalin-fixed, FFPE tumour blocks using the miRNeasy Kit® QIAGEN™. We then performed RNA sequencing using the Archer® technique with the Fusion Plex Gene Panel® Lung V2 (Table S2). This technique, which uses Unique Molecular Identifiers during initial reverse transcription followed by DNA amplification, enables precise detection of variants with very low allelic frequencies while correcting PCR and/or sequencing errors. Quality control of the DNA synthesised during this first stage is carried out by qPCR to eliminate samples whose quality could not provide a contributory molecular analysis. For samples of sufficient quality, the next step in preparing the library for sequencing consisted of amplification by Anchored Multiplex PCR, using primers specific to the fusion genes being sought, as well as universal primers to determine any fusion partner. Finally, sequencing was performed using a Miseq™ sequencer (Illumina®, USA) and fastq files were analysed by Archer Analysis software®.
Exploratory transcriptome study
Our research group has previously published a molecular signature based on transcriptome data from resected PDAC. The data had been obtained following RNA extraction from FFPE tumour resection specimens. We had RNA seq data from several patients with KRAS WT tumours (i.e. the PRODIGE 24 cohort). Using the method described in Puleo et al., we compared expression data between KRAS WT (n = 34) and KRAS-mutated groups (n = 316).
Statistical analysis
Categorical variables are presented as numbers and percentages (%), and continuous variables as median (interquartile range, IQR). Quantitative data were compared using Wilcoxon’s matched pair signed-rank test, and qualitative data were compared using the Chi-squared test and Fisher’s exact test when appropriate. OS was defined as the period between the date of surgery for resected tumours or date of diagnosis of metastases for metastatic tumours, and death from any cause. RFS for resected patients was calculated from the date of surgery to the first recurrence or death (from any cause). Survival curves were estimated using the Kaplan–Meier technique and compared using the log-rank test. For each test, statistical significance was set at a two-tailed p-value of <0.05. All statistical analyses were carried out using the R software.
The reporting of this study conforms to the REMARK statement 33 (Table S3).
Results
KRAS mutations
In total, 929 patients with resected PDAC for whom KRAS status could be determined from tumour material were included. The distribution of tumours by cohort was as follows: Puleo et al. cohort n = 431, Zhao et al. cohort n = 148 and Conroy et al. cohort n = 350. Sequencing data were of low quality for 13 patients due to FFPE artefacts and low coverage (Figure 1(a)). In total, 86/916 patients (9.4%) of resected tumours were KRAS WT. In total, 830 tumours had a KRAS mutation with the following distribution: G12D (n = 198; 38%), G12V (n = 181; 35%), G12R (n = 88; 17%), Q61H (n = 24; 4.6%), G12C (n = 10; 1.9%), Q61K/L/R (n = 7; 1.4%), A59T (n = 2; 0.4%) and other (n = 8; 1.5%). Among KRAS-mutated tumours, the KRAS mutation type was not available for 312 patients (Figure 1(b)).
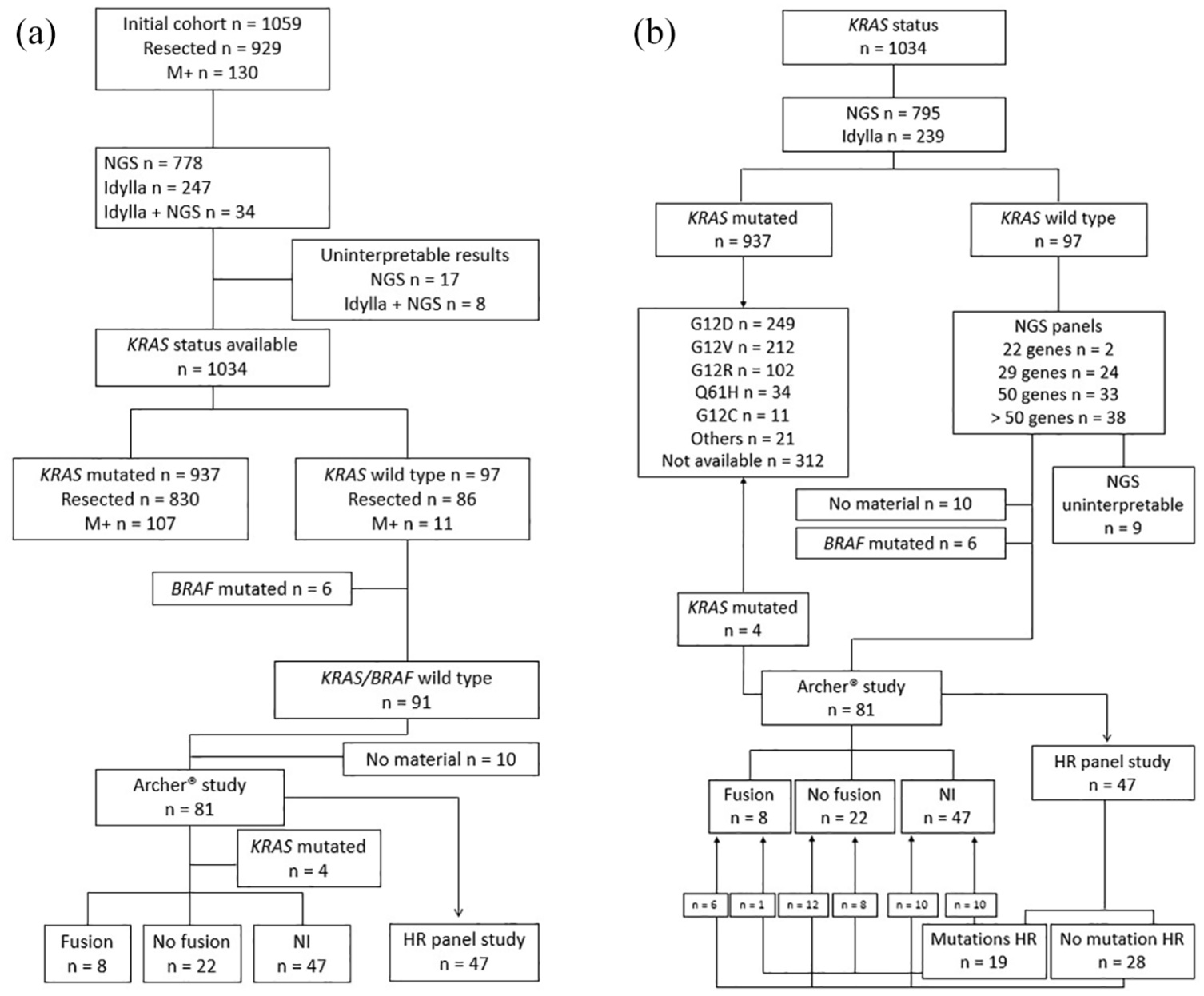
Flow chart. (a) Cohort and molecular analyses. (b) Methods for determining KRAS mutations, fusion genes and other mutations.
In all, 130 patients with a metastatic tumour for which KRAS status could be determined were included. The distribution of tumours by cohort was as follows: monocentric cohort n = 13, PRODIGE 35 cohort n = 85 and PRODIGE 37 cohort n = 32. Sequencing identified 11/118 patients (9.3%) with a KRAS WT metastatic tumour. KRAS status was not interpretable in 12 tumours due to poor quality sequencing (Figure 1(a)). In all, 107 tumours had a KRAS mutation, with the following distribution: G12D (n = 50; 46.7%), G12V (n = 30; 28%), G12R (n = 13; 12.1%), Q61H (n = 9; 8.4%), G12C (n = 1; 0.9%), A59T (n = 1; 0.9%), Q61R (n = 2; 1.8%) and V14I (n = 1; 0.9%; Figure 1(b)).
A total of 97/1034 tumours were KRAS WT, that is, 9.4% of the population, with an equivalent proportion of KRAS WT tumours between the resected and the metastatic group.
Molecular characteristics of KRAS WT tumours
Associated mutations
The 97 KRAS WT tumours underwent NGS to identify the presence of other mutations. The results could not be interpreted for nine tumours. The most frequently found mutation was a mutation in the TP53 gene (n = 24/88, 27.2%). Among the theragnostic mutations of interest, we identified mutations in the BRAF gene. Mutations are detailed in the Supplemental Data (Figure S1). A BRAF mutation was observed in six tumours (6.9% of the KRAS WT population), including 4/78 resected tumours (5% of this population) and 2/10 metastatic tumours (20% of this population). Among BRAF mutations, we found four ‘class 1 mutations’: three ‘V600E mutations’ and one ‘V600R mutation’; as well as two ‘class 3 mutations’: D594N and G466V.
Fusion genes
Gene fusions were assessed by RNA sequencing on 81 KRAS WT/BRAF WT tumours with available material (Figure 1).
Sequencing failed for more than half of the cohort (n = 47), probably secondary to nucleic acid degradation linked to the delay between formalin fixation and RNA extraction. Our samples dated from 1996 to 2016. The older the samples had the higher the failure rate (Table 1).
Failure and success rates of Archer® sequencing analysis according to the age of tumour samples.

Variables are presented as a number (%).
Of the non-mutated samples that were interpretable (n = 34), we identified four KRAS-mutated tumours (G12D n = 1, G12V n = 1, G12R n = 1 and Q61H n = 1), which were therefore excluded. Finally, of the 30 KRAS WT tumours with interpretable Archer sequencing, 22 were fusion free (73%) and 8 were fusion positive (27%; Figure 1). The fusions were mutually exclusive and involved the following genes: NTRK3 (n = 3), NRG1 (n = 2), BRAF (n = 2) and ALK (n = 1). Fusion partners are detailed in the Supplemental Data (Table S4).
Mutations in genes of the homologous recombination pathway
Among the gene panels used for the sequencing of the initial DNA, some were wider and included genes from the homologous recombination (HR) pathway. Of the 77 tumours KRAS WT tested in Archer, 47 tumours were able to benefit from these panels. In all, 19 mutations involving HR pathway genes were identified (n = 19/47, 40%). It should be noted that eight mutations were identified in tumours without a fusion gene, whereas only one tumour with a fusion gene carried a mutation in the HR pathway genes. One of the tumours carrying a fusion no longer had the material required for sequencing the genes of the HR pathway (Figure 1(b)).
Clinical and histological features
The median age of patients with KRAS WT tumours was 62 years (IQR (53–68)). There were statistically more men with a KRAS WT as compared to a KRAS-mutated tumour (66% vs 54%, p = 0.033). The clinico-biological and histological characteristics according to the stage of the disease are detailed in Tables 2 and 3.
Characteristics of the population with a resected KRAS WT tumour.

Variables are presented as a number (%) or median (interquartile range).
WT, wild type.
Characteristics of the population with metastatic KRAS WT tumours.
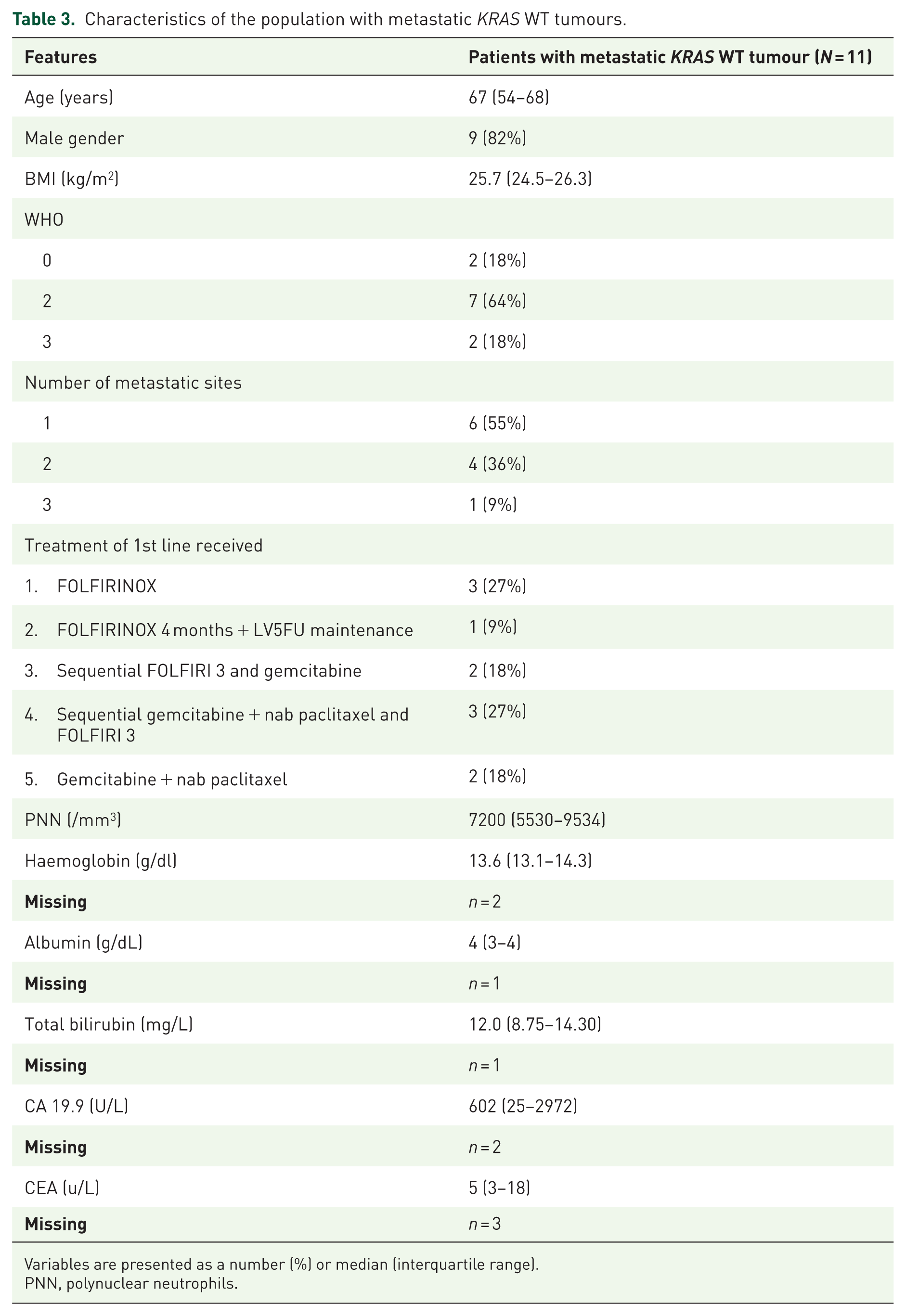
Variables are presented as a number (%) or median (interquartile range).
PNN, polynuclear neutrophils.
OS and RFS according to KRAS status were not statistically different in either the metastatic or resected tumour populations (Figure 2). In exploratory analyses, we compared survival according to KRAS mutations and found no significant difference in either the resected or metastatic tumour groups (Figure S2). We also looked at the correlation between treatment received and KRAS mutation type among metastatic tumours, and found no obvious difference, subject to the small numbers in each subgroup (Table S5).

Survival curves. (a) Recurrence-free survival according to KRAS status in patients with resected tumours. (b) OS according to KRAS status in patients with resected tumours. (c) OS according to KRAS status in patients with metastatic tumours.
Transcriptomic analyses
To evaluate the impact of KRAS mutation on tumour phenotype, gene expression profiles from the PRODIGE24 cohort were used to compare 34 KRAS WT to 316 KRAS-mutated PDAC. Among 3350 differentially expressed genes, 2160 and 1190 were downregulated and upregulated in KRAS WT tumours, respectively. This revealed an overall upregulation of the basal-like or squamous-like phenotype and basal-associated pathway (e.g. keratinisation, interferon signalling) and the enrichment in TGFB-driven cancer-associated fibroblasts in KRAS mutated. On the other hand, KRAS WT tumours tended to show a more immunogenic profile, less activated fibroblasts and displayed a specific well-differentiated molecular profile referred to as Classical-B in a previous study. 34 Overall, these results demonstrate the strong impact of the KRAS mutation in both the epithelial and stromal compartments of PDAC (Figure 3).

Transcriptomic analysis. (a) KRAS pathways. (b) Volcano plot representing differential gene analysis.
Discussion
KRAS WT PDAC is noteworthy for its enrichment in theranostic molecular abnormalities, potentially paving the way for new therapeutic targets. These targets are crucial to optimise the management of these poor-prognosis cancers, with few therapeutic options (frequently at a therapeutic deadlock). Clinical, histological and molecular characterisation of this subgroup of PDAC was poorly described, given the low proportion of KRAS WT tumours and the difficulty in obtaining large quantities of good-quality tumour material, particularly for metastatic and locally advanced tumours. A few large cohorts already exist in the literature. In 2019, Singhi et al. 19 carried out a genomic analysis of more than 3000 PDAC, including 445 KRAS WT tumours, to identify potential markers and therapeutic targets. In 2022, Philip et al. published a molecular characterisation of 266 KRAS WT tumours. They obtained genomic, transcriptomic and clinical data, constituting the best described cohort of KRAS WT tumours to date. 35 Finally, Ashok Kumar et al. 36 recently published a cohort of 9444 cases of locally advanced pancreatic tumours, including 721 KRAS WT cases, to specify the target mutations and their different proportions among KRAS-mutated and non-mutated tumours.
We identified a cohort of 93 KRAS WT tumours from an initial cohort of 929 resected and 130 metastatic cancers. We identified 9% of KRAS WT, with an equivalent proportion between resected and metastatic tumours in line with previous published series. In mutated cases, KRAS G12D was the most frequent alteration, as expected in PDAC. 15 PDAC genotyping may be challenging for patients with metastatic disease, as samples are often very small or have a low tumour cell percentage. Here, KRAS WT samples were validated using NGS, which has a lower detection threshold compared to targeted strategies. Despite the heterogeneity of our cohorts, KRAS genotyping was contributive for most cases. However, four KRAS WT samples were positive for a KRAS mutation by RNA analysis, possibly due to a higher sensitivity of RNAseq to detect highly expressed mutant alleles in a low tumour cell content sample. For those, no fusions were identified.
Eighty-one KRAS WT tumours in our cohort underwent fusion gene testing using the Archer method. Various techniques are available for fusion gene research, such as immunohistochemistry or fluorescence in situ hybridisation, which are still widely used in haematology or thoracic oncology to screen for certain fusions. However, these techniques only allow a sequential search for fusions and require knowledge of the gene of interest without being able to identify the fusion partner. Given these limitations, we chose to use the Archer technique, which utilises RNA sequencing to search for several fusions simultaneously, to determine the location of the chromosomal breakpoint and to identify any fusion, including the partner, from a panel of predefined genes. However, this technique is sensitive to the quality of the RNA. 37 Our samples dated from 1998 to 2016 and had poor quality nucleic acids, causing reverse transcription to fail and making sequencing uninterpretable for more than half of them (58%). Storage techniques, staining and the length of time tumour blocks are responsible for RNA fragmentation and nucleic acid degradation. 38 This generates poor-quality sequencing data that cannot be used for the analysis of gene expression and mutations.
RNA and DNA sequencing data nevertheless revealed several targets of therapeutic interest. Firstly, we identified 8 fusions out of 30 interpretable analyses (27%). The fusion proteins are abnormally active, leading to cellular disruption and the development of cancerous disease. By specifically blocking the fusion protein, some molecules have already proved effective in controlling tumours. For the fusions we have identified, we can mention larotrectinib and entrectinib for NTRK fusion,8,39,40 zenocutuzumab for NRG1 fusion, 7 ALK inhibitors for ALK fusion (crizotinib, ceritinib and alectinib),41,42 and BRAF or MEK inhibitors for BRAF fusion.43–46 We identified six BRAF mutations, that is, 7% of the population of KRAS WT tumours. This proportion is lower than that found in the other cohorts (13% and 17% in Philip’s and Ashok Kumar’s cohorts, respectively), possibly due to a lack of numbers, but remains higher than that known in the overall population of PDAC (3%). 47 The identification of such mutations could, like other cancers such as melanoma or colon cancer, allow the use of BRAFi or MEKi treatment.44–46,48 Finally, we identified a significant proportion of mutations in the genes of the HR pathway (N = 19/47, 40%). It is now accepted that tumours with a BRCA mutation are more sensitive to platinum-based therapy and respond to PARP inhibitors. 49 Although we did not compare these results with KRAS-mutated tumours, Ashok Kumar et al. found a significant difference in mutations, particularly in the ATM gene, between KRAS-mutated and non-mutated tumours without a significant difference for BRCA1 and BRCA2 genes. These results, therefore, do not allow us to state whether a mutation in a gene of the HR pathway is a sufficient condition to lead to oncogenesis. In total, out of the cohort of 93 KRAS WT patients, we identified 33 tumours with at least 1 molecular anomaly of therapeutic interest. This proportion was probably underestimated due to a large number of uninterpretable results.
Concerning the clinical characteristics of patients, we could not identify any phenotype related to KRAS status besides more men in the KRAS WT group (p = 0.03). We did not identify any differences between KRAS-mutated and KRAS WT tumours, whatever the stage of the disease. It has been described in the literature that young patients (<50 years) have a higher incidence of KRAS WT tumours, 50 but the median age of our cohort was in line with the age usually found in patients with PDAC. Some studies have described an improved prognosis in patients with KRAS WT tumours treated with FOLFIRINOX compared with those with KRAS mutation.25,35,51,52 It should be noted that this difference in survival was demonstrated in patients with metastatic disease in the article by Philip et al. Our cohort was mainly constituted of resected tumours, and there were few metastatic tumours, which may have led to a lack of power in this population. Furthermore, the improved prognosis of patients with KRAS WT tumours treated with FOLFIRINOX could be due to the presence of mutations responsible for an HR deficiency, making them more sensitive to platinum-based treatments. 49 The influence of this confounding variable could result in the misleading perception that patients with KRAS WT tumours exhibit improved survival rates, when, in reality, this improvement is associated with mutations in the genes responsible for HR. Adding a transcriptomic analysis for part of our cohort has enabled us to clarify the strong impact of the KRAS mutation on the tumour phenotype, in both the epithelial and stromal compartments. Our results showed that KRAS WT have a different phenotype as compared to KRAS-mutated tumours, and that this molecular profile appears to have a better prognosis, according to the data from Puleo’s study. Our results also align with those of Chan-Seng-Yue et al. 34 that a genomic event establishes molecular subgroups. However, these results need to be confirmed on a larger scale.
Our work has several limitations. Firstly, the retrospective nature of the clinical, histological and sequencing data is responsible for an inevitable bias and missing data. We found the presence of molecular anomalies of therapeutic interest in the KRAS WT population, but the lack of comparison between the KRAS-mutated population and KRAS WT means that we are unable to determine whether there is a significant difference between these two populations, which could guide the diagnostic and then therapeutic strategy. Finally, the age of our samples led to a significant loss of sequencing data due to uninterpretable results, thus reducing our study’s power for fusion gene analyses.
Conclusion
In conclusion, we did not identify any clinical feature that would help us to identify the population with KRAS WT PDAC, and we did not find any difference in prognosis with standard chemotherapy compared with KRAS-mutated tumours. We confirmed that KRAS WT tumours constitute a heterogeneous population from a molecular point of view and are enriched in molecular anomalies of therapeutic interest. These include fusion genes and mutations in genes of the HR pathway, with the potential to target these alterations and improve the prognosis of patients. Our results do not confirm that these abnormalities are found exclusively in KRAS WT tumours, as we did not search for them in the KRAS-mutated population. Nevertheless, we are convinced that screening beyond KRAS for patients with KRAS WT tumours is a chance to access a specific treatment and improve the prognosis of patients with PDAC.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251339939 – Supplemental material for Characteristics of KRAS WT pancreatic adenocarcinomas: results of a large French multicentric cohort
Supplemental material, sj-docx-1-tam-10.1177_17588359251339939 for Characteristics of KRAS WT pancreatic adenocarcinomas: results of a large French multicentric cohort by Noémie Trystram, Jérome Cros, Hélène Blons, Simon Garinet, Rémy Nicolle, Delphine Le Corre, Shu Lin Zhao, Jim Biagi, Alexandre Harlé, Thierry Conroy, Vinciane Rebours, Julien Taïeb, Laetitia Dahan, Pierre Laurent-Puig and Jean-Baptiste Bachet in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.