
Abstract
Background:
Xevinapant, an inhibitor of apoptosis protein (IAP) inhibitor, has shown promising activity in combination with anticancer agents, including radiotherapy, and, in preclinical studies, anti-PD-(L)1 antibodies. This, in part, is due to its ability to restore apoptosis and increase antitumour immunity.
Objectives:
We report efficacy, safety and exploratory biomarker analyses of xevinapant plus avelumab (anti-PD-L1) in a two-part, open-label, nonrandomised, phase Ib study.
Design:
Part A assessed patients with advanced solid tumours who received xevinapant (100, 150, 200 or 250 mg/day, with no random allocation, on Days 1–10 and 15–24) in combination with avelumab (10 mg/kg) on Days 1 and 15 in 28-day cycle. Part B assessed patients with advanced non-small-cell lung cancer (NSCLC) who received xevinapant at the recommended phase II dose (RP2D) plus avelumab (maximum 26 cycles).
Methods:
Part A assessed the safety and tolerability of the combination and established the maximum tolerated dose (MTD) and RP2D of xevinapant. Part B assessed the antitumour activity of xevinapant at the RP2D combined with avelumab compared with a historical control (avelumab alone). Exploratory biomarker analyses were also conducted.
Results:
In part A (n = 16), xevinapant 200 mg/day was established as the RP2D with avelumab and the MTD was not reached. The most common treatment-emergent adverse events (TEAEs) irrespective of xevinapant dose were nausea and fatigue (n = 11 (68.8%) each). In part B (n = 38; four patients received prior anti-PD-(L)1 antibody), the objective response rate (ORR) was 10.5% (95% confidence interval (CI), 2.9–24.8; partial response, n = 4) and the most common TEAE was decreased appetite (n = 13 (34.2%)). Levels of plasma IL-10, IL-1β, IL-13 and CD8+ T cells increased during the study, and circulating levels of CD4+ T cells and Tregs increased during cycle 1. Macrophage-related gene expression signatures increased in patients with a partial response or stable disease. Low baseline Ki-67 expression in tumour samples correlated with a partial response.
Conclusion:
The RP2D of xevinapant with avelumab was established; however, the ORR was not superior to the historical control (avelumab alone). The combination had a manageable safety profile in both study parts. Biomarker analyses provide insights into drivers associated with efficacy in patients with NSCLC receiving xevinapant plus avelumab.
Trial registration:
NCT03270176 (https://clinicaltrials.gov/study/NCT03270176). Registered on ClinicalTrials.gov on 29 August 2017.
Introduction
Lung cancer is the most commonly diagnosed cancer globally, with 2,480,301 new cases and 1,817,172 deaths recorded in 2022. 1 Most cases (≈80%) are non-small-cell lung cancer (NSCLC), 2 and ≈60% of these cases are diagnosed at an advanced stage. 3 Until 2015, chemotherapy and, for a minority of patients, targeted therapies were the most effective options; however, in many cases, de novo resistance or the rapid development of acquired resistance ensued. This mandated the development of novel treatment options. 4 Recently, several phase III clinical trials of anti-programmed cell death 1 (ligand 1) (PD-(L)1) antibodies have shown improved outcomes in patients with advanced NSCLC.5–10 These trials have led to approvals that have changed the treatment landscape for patients with advanced NSCLC.11–13 Treatment with anti-PD-(L)1 antibody alone or with platinum-based chemotherapy has emerged as the standard of care in the first-line setting for patients with tumours that do not have an oncogenic driver. The anti-PD-L1 antibody avelumab is approved in various tumour types, 14 including urothelial carcinoma, Merkel cell carcinoma and renal cell carcinoma. However, in two phase III trials, avelumab did not significantly improve outcomes in patients with NSCLC in the first- or second-line setting.15,16
Inhibitor of apoptosis proteins (IAPs) are a class of proteins that are overexpressed in cancer cells17–19; the median expressions of cellular IAP 1 (cIAP1), cellular IAP 2 (cIAP2) and X-linked IAP (XIAP) in tumour cells from patients with NSCLC have been shown to be 70%, 45% and 25%, respectively. 20 IAPs promote cancer cells’ evasion of apoptosis (a key hallmark of cancer) by inhibiting caspase signalling pathways induced by intrinsic or extrinsic factors and suppressing immune cell activation and antitumour activity.21–25 Xevinapant is a potent, oral, small-molecule IAP inhibitor that is capable of restoring cancer cell sensitivity to apoptosis via a proposed dual mechanism of action (Figure 1), thereby enhancing the effects of chemotherapy and radiotherapy.26–28 Inhibition of XIAP and cIAP1/2 by xevinapant reinitiates caspase activity in the intrinsic and extrinsic apoptotic pathways.26,29 Inhibition of cIAP1/2 increases antitumour immunity by promoting inflammatory cytokine release and switching cytokine signalling from survival to death in the tumours.29–33 This involves increasing proapoptotic tumour necrosis factor (TNF) receptor signalling via the extrinsic pathway and inducing TNF-α expression via the noncanonical nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) pathway as well as inducing receptor-interacting serine/threonine kinase 1- and caspase 8-dependent apoptosis following the inhibition of cIAP1/2.26,34–36 In addition, the inhibition of cIAP1/2 stabilises NF-κB-inducing kinase, resulting in activation of the noncanonical NF-κB pathway and transmission of costimulatory maturation and activation signals to immune cells.37–40 The combination of IAP inhibitors with anti-PD-(L)1 antibodies has shown promising activity in several preclinical cancer models.41–43 Given xevinapant’s novel mechanism of action, with a potential effect on immune cells and promising preclinical activity in combination with anti-PD-(L)1 antibodies, we hypothesised that the combination with avelumab could further enhance antitumour activity.

Xevinapant proposed mechanism of action.
Here, we report topline efficacy and safety results and exploratory biomarker analyses from a phase Ib dose-finding study investigating xevinapant in combination with avelumab in patients with solid tumours and, in the expansion cohort, patients with advanced NSCLC who had disease progression after one line of platinum-based chemotherapy.
Methods
Study design
This open-label, nonrandomised, multicentre, phase Ib dose-finding study (NCT03270176) was divided into two parts. Part A assessed the safety and tolerability of xevinapant in combination with avelumab and the establishment of the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of xevinapant in patients with solid tumours. Part B evaluated the antitumour activity of xevinapant at the RP2D in combination with avelumab in patients with NSCLC who had disease progression after one line of platinum-based chemotherapy. The design in part A used the modified continual reassessment method (mCRM). This method used a dose-toxicity model and a target toxicity rate to estimate the MTD. The first cohort of patients received a starting dose of xevinapant 100 mg/day, and any dose-limiting toxicities (DLTs) were reported to determine the probability of toxicity at each dose and recommend the dose level for the next cohort. The DLT period was defined as the first treatment cycle (i.e. 4 weeks or longer in the event of dosing delays).
Patients received oral xevinapant on Days 1–10 and 15–24 plus intravenous avelumab (10 mg/kg) on Days 1 and 15 of a 28-day cycle. In part A, four dose levels of xevinapant (100, 150, 200 and 250 mg/day) were assessed. In part B, xevinapant was given at the RP2D that was established in part A (200 mg/day on Days 1–10 and 15–24; details described in the ‘Results’ section). Study treatment was planned for 26 cycles or until any of the following: disease progression, permanent discontinuation of avelumab, unacceptable toxicity, patient withdrawal, pregnancy, any medical condition that may risk the patient’s safety if they continued in the study, the start of subsequent anticancer therapy or DLT that did not resolve to a grade ⩽2 within 14 days of onset (part A only). The reporting of this study conforms to the guidelines for reporting nonrandomised studies. 45
Patients
In part A of the study, eligible patients had advanced solid tumours and were either deemed ineligible to receive or had failed standard treatment. In part B, eligible patients had stage IIIB or IV NSCLC and had disease progression after one line of platinum-based chemotherapy. Other key inclusion criteria across both parts included age ⩾18 years; measurable disease per Response Evaluation Criteria in Solid Tumours (RECIST) 1.1; adequate haematological, renal and hepatic function; Eastern Cooperative Oncology Group performance status of 0 or 1 and life expectancy of ⩾3 months. Key exclusion criteria included prior receipt of any immune checkpoint inhibitor treatment (part A), prior receipt of more than one line of anti-PD-(L)1 treatment (part B), prior IAP inhibitor treatment and inability to swallow or retain oral xevinapant.
Endpoints and evaluations
The primary endpoints were as follows: in part A, MTD of xevinapant with the probability of a DLT of <30% and RP2D of xevinapant with avelumab, taking into account the overall cumulative safety, tolerability, pharmacokinetics and efficacy data for the combination; in part B, investigator-assessed objective response rate (ORR) per RECIST 1.1 in a larger cohort of patients with advanced or metastatic NSCLC after platinum-based therapy. Secondary endpoints included treatment-emergent adverse events (TEAEs) and efficacy (best overall response, duration of response, disease control rate, progression-free survival and overall survival) and pharmacokinetics (data not shown). TEAEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03. The final analysis was conducted after the last patient had discontinued from study treatment or had completed the 26 planned treatment cycles. Patients deriving a clinical benefit after 26 cycles were eligible to continue treatment.
Exploratory biomarker analyses were conducted and included analysis of plasma cytokines using multiplex assay (using the Molecular and Cellular Immunology Core, BioCanRx core facility), analysis of circulating immune cells using flow cytometry, immunohistochemistry (IHC) and NanoString gene expression analyses (using the nCounter PanCancer Immunology 360 panel (NanoString Technologies, Seattle, WA, USA)) on RNA extracted using the RecoverAll™ Total Nucleic Acid Isolation Kit for FFPE from pretreatment tumour samples. Activated regulatory T cells (Tregs) assessed by flow cytometry and NanoString analyses were characterised as FOXP3+. The biomarkers analysed in the study and the stains used for flow cytometry parameters are presented in Supplemental Table 1. IHC slides were incubated at 37°C overnight, deparaffinised in xylene and rehydrated in graded ethanol baths (Fisher BioReagents, ThermoFisher, Waltham, MA, USA). Antigen retrieval was performed using Diva Decloaker (Biocare Medical, LLC™, Pacheco, CA, USA) in a Decloaking Chamber (Biocare Medical, LLC). Slides were stained using the intelliPATH FLX® (Biocare Medical, LLC) with the following panels: CD8/CD3/PanCK, cIAP1/PanCK/Ki-67, FOXP3/PanCK/CD68 and PD-L1/PD-1/CD8. Slides were imaged using the Vectra 3 Automated Quantitative Pathology Imaging System (Akoya Biosciences, Inc™, Marlborough, MA, USA) and analysed using the inForm Tissue Analysis Software (Akoya Biosciences, Inc™). For NanoString gene expression counts, immune cell scores were calculated by the average log2 expression of all cell type-specific markers (Supplemental Table 2); the corresponding cell type-specific genes that were used in this study were previously described by Danaher et al. 46
Statistical analysis
The statistical analyses were descriptive in part A. In part B, the primary endpoint was compared against a historical ORR of 10% achieved with avelumab monotherapy. 47 The null hypothesis was that there was no difference in the ORR between the treatments in this study (xevinapant and avelumab) and the historical control (avelumab). The alternative hypothesis was that there was a difference (target ORR, ⩾15%). For an exact binomial test of a proportion with a one-sided nominal significance level of 0.1 and a null proportion of 0.10, a sample size of 35 has an exact power of 0.808 when the true proportion is 0.25. Based on these assumptions, if at least 7 of 35 evaluable patients had an objective response, the study would be deemed positive.
Test statistics and p values were derived from different models for each biomarker analysis. p Values for cytokines were derived from the linear mixed-effect models with the p value corresponding to the fixed effect of the ordered categorical visit. p Values for IHC analyses of tumour samples and NanoString immune cell scores were derived from different regression models depending on the clinical efficacy endpoint with a logistic model for binary endpoints and an ordinal logistic model for multilevel categorical endpoints. p Values < 0.05 were considered statistically significant, with no adjustment for multiple comparisons.
Results
Results from part A
Between October 2017 and March 2021, 19 patients were screened. A total of 16 patients with solid tumours (lung, n = 5 (31.3%); ovary, n = 2 (12.5%); kidneys/adrenals, n = 1 (6.3%); stomach, n = 1 (6.3%); other, n = 7 (43.8%)) were enrolled at four centres in Canada and treated across the four xevinapant dose levels (100 mg/day, n = 3; 150 mg/day, n = 2; 200 mg/day, n = 7; 250 mg/day, n = 4). A maximum of 26 cycles of treatment was received by 2 patients (12.5%); 14 patients (87.5%) discontinued treatment due to disease progression (11 (78.6%)) and adverse events (AEs; 3 (21.4%)). The median age was 59.5 years (range, 28–79 years); 50% were male and 50% were female (n = 8 each; Table 1).
Baseline characteristics for patients in part A and part B.

Percentages may not total 100 because of rounding.
Pleura in two patients, and colorectal, duodenal, soft tissues of the abdomen, thymus and thyroid in one patient each.
Adrenal cortical carcinoma, epithelioid mesothelioma, leiomyosarcoma, malignant epithelial mesothelioma, papillary and serous carcinoma in one patient each.
Large-cell carcinoma in two patients, and mucinous bronchioloalveolar carcinoma, pulmonary carcinosarcoma and NSCLC (not otherwise specified) in one patient each.
Assessed using the PD-L1 IHC 22C3 pharmDx assay. PD-L1 expression was deemed to be high when PD-L1 was observed in ≥50% of tumour cells and moderate when PD-L1 was observed in 1%−49% of tumour cells.
ECOG PS, Eastern Cooperative Oncology Group performance status; NA, not analysed; NSCLC, non-small-cell lung cancer; PD-(L)1, programmed cell death 1 (ligand 1).
The MTD of xevinapant was not reached. DLTs were reported for one patient at the 250-mg/day dose level (reversible grade 3 alanine aminotransferase (ALT) and aspartate aminotransferase (AST) increases). The safety monitoring committee endorsed the recommendation of the mCRM, establishing xevinapant 200 mg/day on Days 1–10 and 15–24 every 28 days as the RP2D based on the cumulative safety, efficacy and pharmacokinetic (data not shown) parameters. The dose of 200 mg/day was selected due to the observed DLT in a patient at the 250-mg/day dose level, as well as the occurrence of grade 3 myalgia and arthralgia in a patient at the 200-mg/day dose level which were considered to be related to both study treatments.
No increases to grade ⩾2 in ALT or AST were observed at lower dose levels. TEAEs occurred in all patients, and grade ⩾3 TEAEs were observed in 10 patients (62.5%; Table 2). The most common TEAEs irrespective of xevinapant dose were nausea and fatigue, both occurring in 11 patients (68.8%; Supplemental Table 3); fatigue was also the most common grade ⩾3 TEAE irrespective of xevinapant dose (n = 3 (18.8%); Supplemental Table 4). TEAEs that resulted in permanent treatment discontinuation were reported in three patients (grade 3 increase in AST levels and grade 3 increase in ALT levels, grade 2 pneumonitis and grade 3 arthralgia, respectively). The most common TEAEs related to study treatments are presented in Supplemental Table 5 (TEAEs related to xevinapant) and Supplemental Table 6 (TEAEs related to avelumab). Grade 3 TEAEs related to xevinapant and/or avelumab were observed in five patients (31.3%). No patients died during the study.
Safety results from part A and part B.

TEAE, treatment-emergent adverse event.
Efficacy and safety results from part B
Between May 2019 and March 2022, 48 patients were screened. Overall, 38 patients with NSCLC were enrolled and treated at six centres in Canada, Poland and Romania. Baseline characteristics of all 38 patients enrolled in part B are presented in Table 1. All patients had received prior chemotherapy and 10.5% of patients had received prior therapy with an anti-PD-(L)1 antibody. As of March 2022, 37 patients (97.4%) had discontinued study treatment early and 1 patient (2.6%) had completed the planned 26 cycles of treatment (Supplemental Figure 1). The median number of cycles of xevinapant and avelumab was 4 (range, 1–26). The median duration of treatment was 3.6 months (range, 0.1–30.2 months) with xevinapant and 3.7 months (range, 0.2–30.3 months) with avelumab. Of those who discontinued treatment (n = 37), the most common reasons for discontinuation were disease progression (n = 26 (70.3%)) and AEs (n = 10 (27.0%)). PD-L1 expression in tumour samples was high (⩾50%) in 13 patients (34.2%) and moderate (1%−49%) in 5 patients (13.2%); 12 patients (31.6%) had PD-L1-negative disease and PD-L1 status was unknown in 8 patients (21.1%).
All patients had measurable disease and were evaluable for efficacy. Efficacy results from part B are presented in Table 3. The observed ORR was 10.5% (95% CI, 2.9–24.8), with four patients responding to treatment; thus, the target of a ⩾15% increase in efficacy over the historical control (avelumab monotherapy; ORR, 10%) was not met.
Efficacy results from part B.
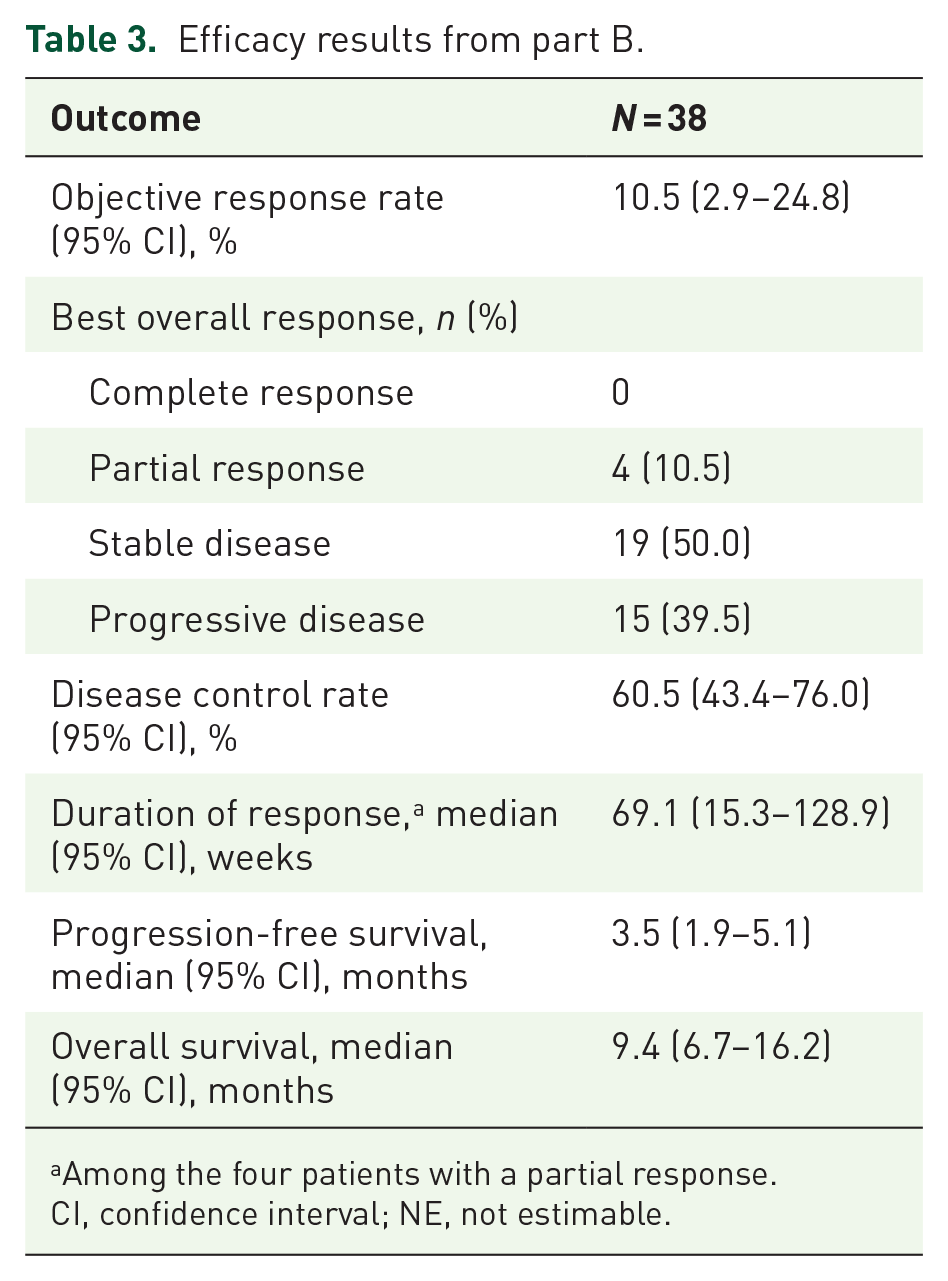
Among the four patients with a partial response.
CI, confidence interval; NE, not estimable.
TEAEs of any grade and from any cause occurred in 37 patients (97.4%; Table 2). The most common TEAEs were decreased appetite (n = 13 (34.2%)), ALT increased (n = 11 (28.9%)), anaemia and amylase increased (n = 10 (26.3%) each; Supplemental Table 7), and the most common grade ⩾3 TEAE was amylase increased (n = 5 (13.2%); Supplemental Table 8). The most common TEAEs related to xevinapant, irrespective of grade, were decreased appetite and nausea (n = 7 (18.4%) each; Supplemental Table 9). TEAEs related to xevinapant were grade ⩾3 in one patient (2.6%; amylase increased). The most common TEAEs related to avelumab, irrespective of grade, were amylase increased (n = 9 (23.7%)) and hypothyroidism (n = 7 (18.4%); Supplemental Table 10). TEAEs related to avelumab were grade ⩾3 in six patients (15.8%; amylase increased in four (10.5%), lipase increased in three (7.9%) and ALT increased in one (2.6%)). No serious TEAEs were considered related to either treatment. Nine patients (23.7%) died during the study (due to disease progression in 7 (18.4%) and pneumonia in 1 (2.6%); cause of death was unknown in 1 (2.6%)).
Biomarker analysis from part B
Significant increases in plasma interleukin (IL)-10 (p = 0.0001; Figure 2(a)), IL-1β (p = 0.0043; Figure 2(b)) and IL-13 (p = 0.0092; Figure 2(c)) levels were detected at all measured time points across the course of the study. Levels of other cytokines, such as IL-12p40 (data not shown), IL-1Ra (data not shown) and TNF-α (Figure 2(d)), also increased at one time point versus baseline; however, this was not maintained across the whole of the study (Table 4). In analyses of peripheral blood, activated CD4+ T-cell and Treg levels increased during cycle 1, and Tregs tended to decrease by Day 1 of cycle 4; activated CD8+ T-cell levels increased throughout the study treatment period versus baseline (Figure 3). Of the four responders (all with a partial response), none had received any prior anti-PD-(L)1 treatment and three had tumours with high PD-L1 expression; PD-L1 status for the fourth patient was not known. In IHC analyses of tumour samples, there was no association between best overall response and PD-L1 expression (p = 0.8383), or between best overall response and Ki-67 expression (p = 0.137), with the four patients with a best overall response of partial response having lower Ki-67 expression at baseline than patients with stable disease or progressive disease (Figure 4). No association was observed between cIAP1 levels and best overall response (p = 0.5541). In NanoString gene expression analyses of tumour samples, high levels of macrophages (p = 0.0248) and Tregs (p = 0.0458) were associated with disease control (Figure 5). All NanoString immune cell score results are presented in Table 5.
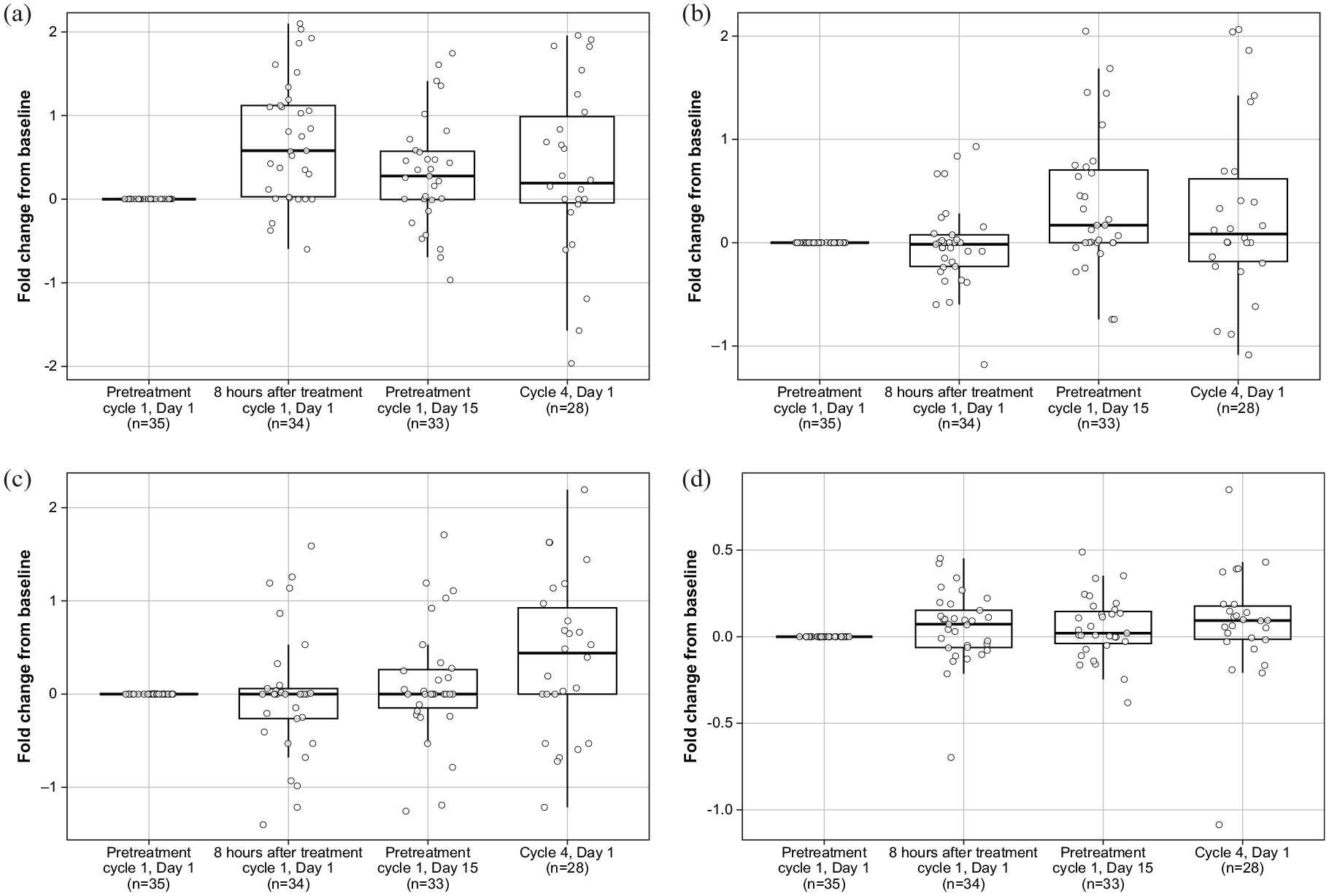
Change in IL-10 (a), IL-1β (b), IL-13 (c) and TNF-α (d) levels throughout the treatment period versus at baseline in part B.
p Values for levels of cytokines in the blood during the study versus at baseline in part B.

GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; TNF, tumour necrosis factor.

Change in levels of activated CD4+ T cells (a), CD8+ T cells (b) and CD4+/CD25+/FOXP3+ Tregs (c) in the blood throughout the study treatment period versus at baseline in part B.

Association between IHC expression of Ki-67 at baseline and best overall response in part B.

Distribution of NanoString immune cell scores for macrophages (a) and FOXP3+ Tregs (b) in patients with and without disease control in part B.
NanoString immune cell scores for all biomarkers from FFPE biopsies during the study versus at baseline in part B.

BOR, best overall response; DC, disease control; FFPE, formalin-fixed paraffin-embedded; NK, natural killer; OS, overall survival; Treg, regulatory T cell.
Discussion
This phase Ib dose-finding study was the first clinical study to evaluate the combination of xevinapant and avelumab or any combination of an IAP inhibitor and anti-PD-(L)1 antibody. In part A, the RP2D of xevinapant in combination with avelumab was selected as 200 mg/day on Days 1–10 and 15–24 of a 28-day cycle based on a manageable safety profile. Reversible DLTs (grade 3 ALT and AST increase) were reported for one patient at the 250-mg/day dose level. In part B, xevinapant in combination with avelumab had a manageable safety profile in patients with advanced NSCLC who had disease progression after one line of platinum-based chemotherapy. Incidence and severity of common TEAEs were consistent with the known safety profile of xevinapant and avelumab treatment.48-50 Nevertheless, the study did not meet its primary efficacy endpoint – the addition of xevinapant to avelumab did not improve antitumour activity compared with historical data for avelumab monotherapy as second-line treatment. 51
Comprehensive biomarker data were reported and generally reflected the potential immunomodulatory effects of the combination of xevinapant and avelumab. An increase in CD8+ T-cell expression following xevinapant monotherapy in squamous cell carcinoma of the head and neck (SCCHN) has been reported previously, 35 consistent with our findings in NSCLC. Increased CD8+ T-cell expression has previously been associated with improved survival in several trials of anti-PD-(L)1 antibodies.52–54 The low response rate in part B of the study suggests that this increase from baseline in CD8+ T cells was insufficient to elicit a significant anti-tumour response. As there was no control arm in the study, observed associations may be solely prognostic and independent of treatment. Our data mimic the mechanism of action of xevinapant to some extent as we identified increases in levels of cytokines regulated by NF-κB, consistent with results from preclinical studies,26,35 which have shown that xevinapant is capable of enhancing NF-κB activity. In the JAVELIN Bladder 100 trial, increased expression of genes associated with macrophages correlated with longer overall survival with avelumab, 52 matching the increase in macrophage immune cell scores observed in patients with disease control in our study. However, in the JAVELIN Head and Neck 100 trial in patients with locally advanced SCCHN treated with avelumab plus chemoradiotherapy versus placebo plus chemoradiotherapy, high levels of a signature associated with M0/M2 macrophages correlated with shorter progression-free survival (data not shown). 55 These contrasting findings are caveated by data from other tumour types and differing approaches in measuring cell populations. Circulating levels of IL-10, IL-1β and IL-13 increased during the study, and levels of Tregs increased during cycle 1. Activated Tregs have been shown to stimulate the production of IL-10, 56 and both have been shown to inhibit the antitumour response by restricting the activity of CD4+ T cells; this was also observed in our study, with a reduction in levels of activated CD4+ cells between cycle 1 and cycle 4. These findings could explain the lack of an antitumour response observed in this study. IL-10 expression has also been identified as a poor clinical prognosis factor for NSCLC. 57 Similarly, IL-1β and IL-13 have been shown to contribute to the generation of an immunosuppressive microenvironment across various cancer types.58,59 In patients with NSCLC, IL-1β expression has been shown to correlate with poor efficacy (reduced progression-free survival). 60 The biomarker analyses presented here support the lack of an antitumour response observed with xevinapant and avelumab treatment in our study and indicate an immunosuppressive phenotype. Further studies would be needed to confirm our results and clarify how these biomarker changes inform clinical endpoints.
The findings should be interpreted in the context of a few limitations. The absence of a control arm in part B hinders interpretation of the efficacy and biomarker findings. Furthermore, the biomarker analyses were exploratory, and the patient numbers were limited; therefore, p values should be interpreted with caution. The low number of patients with a response in part B (n = 4 (10.5%), all with a partial response) did not allow for significant associations between biomarker levels and clinical efficacy endpoints. In addition, biopsies were archived samples (not older than 1 year); therefore, biomarker findings should be interpreted with caution. To clarify the role of xevinapant, future studies are needed with larger sample sizes and a control arm, thereby allowing the immune impact of adding xevinapant to be more precisely defined.
Conclusion
Although part B of the phase Ib study failed to meet its primary objective, the combination was safe and feasible, and the biomarker analyses reported here provide novel insights into the drivers associated with efficacy in patients with NSCLC receiving xevinapant in combination with avelumab.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251332154 – Supplemental material for Xevinapant plus avelumab in advanced solid tumours, with a dose expansion in advanced non-small-cell lung cancer: exploratory biomarker, safety and efficacy analyses from an open-label, nonrandomised phase Ib study
Supplemental material, sj-docx-1-tam-10.1177_17588359251332154 for Xevinapant plus avelumab in advanced solid tumours, with a dose expansion in advanced non-small-cell lung cancer: exploratory biomarker, safety and efficacy analyses from an open-label, nonrandomised phase Ib study by Glenwood Goss, Tudor Ciuleanu, Rodryg Ramlau, Daniel J. Renouf, Quincy Chu, Ewa Kalinka, Piotr Sawrycki, Jonathan Bramson, Brad H. Nelson, Rafael Crabbé, Eric LaCasse, Bryan Lo, Daniela A. Sahlender, Philippa Crompton, Franck Brichory, Luke Piggott, Michael Schenker and Rosalyn Juergens in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359251332154 – Supplemental material for Xevinapant plus avelumab in advanced solid tumours, with a dose expansion in advanced non-small-cell lung cancer: exploratory biomarker, safety and efficacy analyses from an open-label, nonrandomised phase Ib study
Supplemental material, sj-docx-2-tam-10.1177_17588359251332154 for Xevinapant plus avelumab in advanced solid tumours, with a dose expansion in advanced non-small-cell lung cancer: exploratory biomarker, safety and efficacy analyses from an open-label, nonrandomised phase Ib study by Glenwood Goss, Tudor Ciuleanu, Rodryg Ramlau, Daniel J. Renouf, Quincy Chu, Ewa Kalinka, Piotr Sawrycki, Jonathan Bramson, Brad H. Nelson, Rafael Crabbé, Eric LaCasse, Bryan Lo, Daniela A. Sahlender, Philippa Crompton, Franck Brichory, Luke Piggott, Michael Schenker and Rosalyn Juergens in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
The authors thank the patients and their families, and the investigators, co-investigators and study teams at each of the participating centres. Avelumab was provided by Merck (CrossRef Funder ID: 10.13039/100009945) and Pfizer. Medical writing support was provided by Jamie Ratcliffe of Nucleus Global and was funded by Merck, in accordance with Good Publication Practice guidelines (![]() ). Merck reviewed the manuscript for medical accuracy. The authors are fully responsible for the content, and the views and opinions described in the publication reflect solely those of the authors. Dr Harman Sekhon of The Ottawa Hospital, Ottawa, ON, Canada, was the pathologist who carried out PD-L1 staining and analyses. Katy Milne of the Deeley Research Centre, BC Cancer – Victoria, Victoria, BC, Canada, was a pathologist who helped to develop the IHC methods and conduct the IHC analyses. Jessica Irvine, Jamie McNicol and Ying Wu helped to collect peripheral blood mononuclear cells and serum, perform flow cytometry and cytokine assessment and analyse the resulting data. Dr Robert Korneluk of the CHEO Research Institute, Ottawa, ON, Canada, contributed to the obtaining the funding for this study.
). Merck reviewed the manuscript for medical accuracy. The authors are fully responsible for the content, and the views and opinions described in the publication reflect solely those of the authors. Dr Harman Sekhon of The Ottawa Hospital, Ottawa, ON, Canada, was the pathologist who carried out PD-L1 staining and analyses. Katy Milne of the Deeley Research Centre, BC Cancer – Victoria, Victoria, BC, Canada, was a pathologist who helped to develop the IHC methods and conduct the IHC analyses. Jessica Irvine, Jamie McNicol and Ying Wu helped to collect peripheral blood mononuclear cells and serum, perform flow cytometry and cytokine assessment and analyse the resulting data. Dr Robert Korneluk of the CHEO Research Institute, Ottawa, ON, Canada, contributed to the obtaining the funding for this study.
Author’s note
Franck Brichory, affiliation at the time the study was conducted.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.