
Abstract
Non-small-cell lung cancer (NSCLC) is a highly heterogeneous disease that is frequently associated with a host of known oncogenic alterations. Advances in molecular diagnostics and drug development have facilitated the targeting of novel alterations such that the majority of NSCLC patients have driver mutations that are now clinically actionable. The goal of this review is to gain insights into clinical research and development principles by summary, analysis, and discussion of data on agents targeting known alterations in oncogene-driven, advanced NSCLC beyond those in the epidermal growth factor receptor (EGFR) and the anaplastic lymphoma kinase (ALK). A search of published and presented literature was conducted to identify prospective trials and integrated analyses reporting outcomes for agents targeting driver gene alterations (except those in EGFR and ALK) in molecularly selected, advanced NSCLC. Clinical efficacy data were extracted from eligible reports and summarized in text and tables. Findings show that research into alteration-directed therapies in oncogene-driven, advanced NSCLC is an extremely active research field. Ongoing research focuses on the expansion of new agents targeting both previously identified targets (particularly hepatocyte growth factor receptor (MET), human epidermal growth factor receptor 2 (HER2), and Kirsten rat sarcoma viral oncogene homolog (KRAS)) as well as novel, potentially actionable targets (such as neuregulin-1 (NRG1) and phosphatidylinositol 3-kinase (PI3K)). The refinement of biomarker selection criteria and the development of more selective and potent agents are allowing for increasingly specific and effective therapies and the expansion of clinically actionable alterations. Clinical advances in this field have resulted in a large number of regulatory approvals over the last 3 years. Future developments should focus on the continued application of alteration therapy matching principles and the exploration of novel ways to target oncogene-driven NSCLC.
Keywords
Introduction
Lung cancer is one of the most common malignancies and the leading cause of cancer-related death.1–3 Non-small-cell lung cancer (NSCLC) accounts for 85% of all lung malignancies and approximately 50% of NSCLC patients are diagnosed at the metastatic stage.4–6
NSCLC is a heterogeneous disease that is frequently associated with multiple known oncogenic driver genes.7–10 The earliest characterized of these are mutations involving the epidermal growth factor receptor (EGFR) and fusions involving anaplastic lymphoma kinase (ALK). Treatment with EGFR and ALK inhibitors is well established, with initial approvals in biomarker-unselected and -selected populations in 2003 and 2011, respectively.7,11–14
Advances in cancer biology, molecular diagnostics, and drug development have improved our ability to identify and therapeutically target oncogenic alterations.9,10,15,16 It is now estimated that the majority of NSCLC patients have alterations that are clinically actionable with a therapeutic agent that acts on the altered target (alteration-drug-matched).9,17–26 Here we will update our initial review of novel (non-EGFR/ALK) targeted therapies 8 by identifying, summarizing, analyzing, and discussing recent data on agents targeting lesser-known alterations (Table 1) in oncogene-driven, advanced NSCLC to gain insights into clinical research and development principles.
Select actionable molecular alterations in oncogene-driven NSCLC.
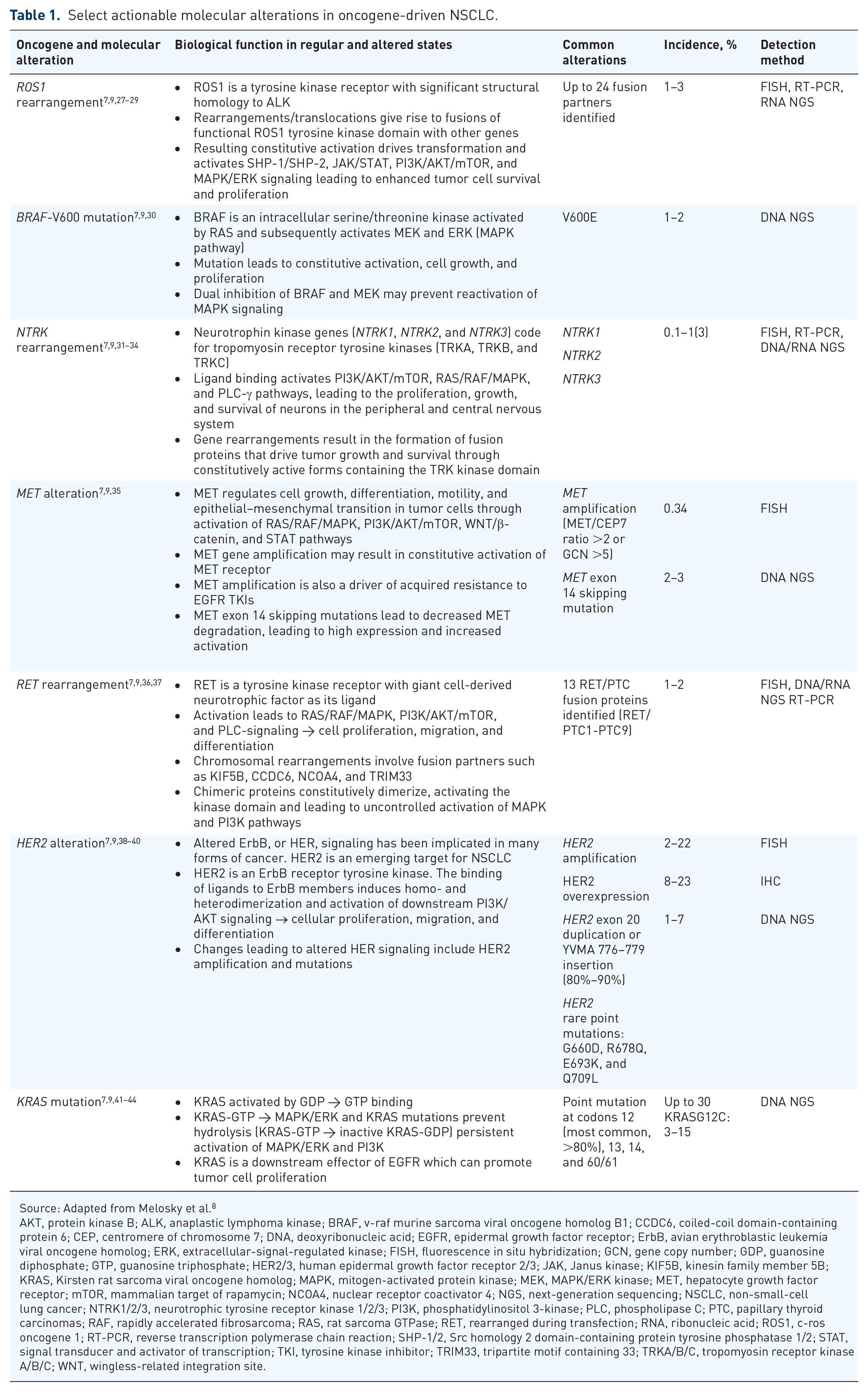
Source: Adapted from Melosky et al. 8
AKT, protein kinase B; ALK, anaplastic lymphoma kinase; BRAF, v-raf murine sarcoma viral oncogene homolog B1; CCDC6, coiled-coil domain-containing protein 6; CEP, centromere of chromosome 7; DNA, deoxyribonucleic acid; EGFR, epidermal growth factor receptor; ErbB, avian erythroblastic leukemia viral oncogene homolog; ERK, extracellular-signal-regulated kinase; FISH, fluorescence in situ hybridization; GCN, gene copy number; GDP, guanosine diphosphate; GTP, guanosine triphosphate; HER2/3, human epidermal growth factor receptor 2/3; JAK, Janus kinase; KIF5B, kinesin family member 5B; KRAS, Kirsten rat sarcoma viral oncogene homolog; MAPK, mitogen-activated protein kinase; MEK, MAPK/ERK kinase; MET, hepatocyte growth factor receptor; mTOR, mammalian target of rapamycin; NCOA4, nuclear receptor coactivator 4; NGS, next-generation sequencing; NSCLC, non-small-cell lung cancer; NTRK1/2/3, neurotrophic tyrosine receptor kinase 1/2/3; PI3K, phosphatidylinositol 3-kinase; PLC, phospholipase C; PTC, papillary thyroid carcinomas; RAF, rapidly accelerated fibrosarcoma; RAS, rat sarcoma GTPase; RET, rearranged during transfection; RNA, ribonucleic acid; ROS1, c-ros oncogene 1; RT-PCR, reverse transcription polymerase chain reaction; SHP-1/2, Src homology 2 domain-containing protein tyrosine phosphatase 1/2; STAT, signal transducer and activator of transcription; TKI, tyrosine kinase inhibitor; TRIM33, tripartite motif containing 33; TRKA/B/C, tropomyosin receptor kinase A/B/C; WNT, wingless-related integration site.
Methods
We have elected to support this narrative review with systematic search methods to ensure unbiased and comprehensive identification, assessment, and summary of relevant clinical studies in this field. A search of published and presented literature was conducted to identify prospective phase I–III trials and integrated analysis reporting efficacy outcomes for agents targeting novel driver gene alterations (i.e., excluding EGFR and ALK) in molecularly selected, advanced NSCLC populations. PubMed (all time to October 25, 2023), the proceedings from the American Society of Clinical Oncology (ASCO), the European Society for Medical Oncology (ESMO), and the World Conference on Lung Cancer 2022 and 2023 annual meetings were searched using the key search terms “NSCLC” AND “advanced”/“metastatic” AND “novel targets” AND “phase I–III” OR respective aliases (Figure 1). A supplemental bibliographic search of review articles and pooled/meta-analyses was also conducted. In addition, directed searches were performed after the database search cutoff date to ensure that the most up-to-date reports of eligible studies were considered. English language records were vetted at the abstract level and checked at the full text as needed by an initial reviewer (A.P.) and confirmed by a second independent reviewer (I.M.). All eligible studies were cited in the findings; however, only trials reporting outcomes since our initial review 8 (i.e., in the last 3 years, approximately) for at least 20 patients with alteration-drug-matched NSCLC were included in the tables. Additional search and vetting details are summarized in Supplemental Methods.

PRISMA diagram.
Findings
The literature search identified a total of 2938 records, resulting in a total of 180 primary reports from 174 clinical trials or integrated analyses reporting efficacy outcomes on novel oncogene-directed therapies in alteration-matched advanced NSCLC (PRISMA, Figure 1). Study analyses were grouped by molecular target and will be discussed chronologically based on the availability of a Food and Drug Administration (FDA) approved agent: c-ros oncogene 1 (ROS1)-rearranged (n = 23), v-Raf murine sarcoma viral oncogene homolog B1 (BRAF) V600-mutated (n = 9), neurotrophic tyrosine receptor kinase (NTRK)-rearranged (n = 4), hepatocyte growth factor receptor (MET)-altered (n = 35), rearranged during transfection (RET)-rearranged (n = 15), human epidermal growth factor receptor (HER)2-altered (n = 38), Kirsten rat sarcoma viral oncogene homolog (KRAS)-mutated (n = 32), fibroblast growth factor receptor (FGFR)-altered (n = 10), HER3-/neuregulin-1 (NRG1)-altered (n = 10), phosphoinositide 3-kinase (PI3K)-altered (n = 2), and tyrosine-protein kinase-like 7 (PTK7)-mutated (n = 1). Analyses with ⩾20 NSCLC biomarker-selected patients reporting outcomes in the last 3 years (since September 2020) are summarized in Table 2 (n = 73).
Efficacy outcomes of clinical trials assessing novel targeted therapy in molecularly selected, target-matched advanced NSCLC.

Study inclusion criteria: Presented or published clinical trials of novel targeted therapy agents assessed in molecularly selected, alteration-drug-matched advanced/metastatic NSCLC. Trials reporting efficacy outcomes in the last 3 years and with ⩾20 NSCLC patients as well as select pivotal trials are included. Trials exclusively in the locally advanced setting were not included. This table serves as an update to Table 2 of our initial review on this topic 8 —studies included in that table were not included here unless their results were updated since the cutoff date for the previous review.
By independent review unless otherwise specified.
By investigator (local radiological) assessment.
Based on the initial report of clinical activity in 26 ROS1+ patients enrolled in the respective study cohort. 48
With a 7-day lead-in period at 90 mg.
Type of radiological assessment (by investigator or independent review) not specified.
CT of investigator’s choice with or without pembrolizumab 200 mg q3w.
Includes two patients still on treatment with partial responses pending confirmation.
Includes unconfirmed responses.
Transformed from weeks to months using a conversion factor of 4.35 weeks/month.
With an updated data cut-off date of January 15, 2022 (median follow-up of 15.6 months).
Patients enrolled in three cohorts according to the type of alteration: High confidence activating FGFR translocations (1), high confidence activating FGFR mutations (2), and low confidence activating FGFR alterations (3).
Both responses were observed in the cohort of patients with high confidence activating FGFR translocations. ORR in that cohort was 29%.
ALK, anaplastic lymphoma kinase; BID, twice daily; BRAF, v-Raf murine sarcoma viral oncogene homolog B1; CEP, centromere of chromosome 7; CI, confidence interval; c-Met, hepatocyte growth factor receptor; CT, chemotherapy; DoR, duration of response; EGFR, epidermal growth factor receptor; FGFR, fibroblast growth factor receptor; GCN, gene copy number; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; HRG, heregulin; IHC, immunohistochemistry; KRAS, Kirsten rat sarcoma viral oncogene homolog; MET, hepatocyte growth factor receptor; mos, months; n, number; NCT, national clinical trial; NE, not estimable; NR, not reported; NRG1, neuregulin-1; NS, non-significant; NSCLC, non-small-cell lung cancer; NSQ, non-squamous cell; NTRK, neurotrophic tyrosine receptor kinase; NYR, not yet reached; ORR, objective response rate; OS, overall survival; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; PFS, progression-free survival; PTK7, tyrosine-protein kinase-like 7; QD, once daily; qXw, every X weeks; R2PD, recommended phase II dose; RET, rearranged during transfection; ROS1, c-ros oncogene 1; SQ, squamous cell; T-DM1, trastuzumab emtansine; T-DXd, trastuzumab deruxtecan; TKD, tyrosine kinase domain; TKI, tyrosine kinase inhibitor; WT, wild-type; #th line+, #th line of treatment in the advanced setting or higher.
ROS1-rearranged
The ROS1 tyrosine kinase domain shares 84% and 86% sequence similarity with ALK and tropomyosin receptor kinase (TRK), respectively.133,134 Consequently, two main types of inhibitors have been assessed in patients with tyrosine kinase inhibitor (TKI)-naïve and -pretreated NSCLC harboring ROS1 rearrangements: those that co-inhibit ALK/ROS153,135–146 and those that co-inhibit TRK/ROS1.144–147 In TKI-naïve patients, the ALK/ROS1/MET co-inhibitor crizotinib was the first agent to receive regulatory approval for NSCLC with ROS1 rearrangements in 2016 based on results from the phase I PROFILE-1001 study. 148 Updated results confirmed initial findings of frequent (objective response rate (ORR) 72%) and durable (median duration of response (mDoR) 24.7 months) responses and showed strong median progression-free survival (mPFS 19.3 months) and median overall survival (mOS 51.4 months) outcomes. 141 Multiple other phase I–II studies of crizotinib in ROS1-rearranged NSCLC have confirmed its clinical efficacy45,46,48,50,140 and updated results from three of these studies demonstrated robust survival outcomes with mPFS of 13.8–19.4 months and mOS of 40.5–54.8 months at a median follow-up of 54.4–81.4 months45,47,49,50 (Table 2). With few exceptions where ORRs were modest (ensartinib 27%, iruplinalkib 36.8%),55,138 other ALK/ROS1 inhibitors (ceritinib, lorlatinib, brigatinib, and unecritinib) have shown high clinical activity as initial ROS1-directed therapy (ORR 62%–80.2%),53,54,136,142,143 however have yet to receive regulatory approval for ROS1-rearranged NSCLC. In particular, unecritinib (a novel derivative of crizotinib) was recently associated with an 80.2% ORR, 20.3-month mDoR, and mPFS of 16.5 months. 53
Entrectinib is a brain-penetrant agent and was the first TRK/ROS1 inhibitor to receive accelerated approval for TKI-naïve ROS1-rearranged NSCLC based on results from an NSCLC-specific integrated analysis of phase I–II studies in multiple tumors. 149 At a median follow-up of 29.1 months, entrectinib continued to show frequent and durable responses (ORR 67.9%; mDoR 20.5 months), with a mPFS and mOS of 15.7 and 47.8 months, respectively. 52 Taletrectinib and repotrectinib are potent, next-generation TRK/ROS1 inhibitors designed to improve central nervous system activity and overcome treatment resistance with favorable safety profiles that showed early signs of activity in ROS1-rearranged NSCLC.146,147,150 Treatment with repotrectinib resulted in frequent and durable responses and robust PFS in the ROS1-TKI-naïve cohort of the phase II trial TRIDENT-1 (ORR 79%; mDoR 34.1 months; mPFS 35.7 months),56,144 leading to a priority review designation 151 and subsequent approval by the FDA. 152 After displaying promising activity in phase I studies,147,153 taletrectinib showed the highest response rates across studies of ROS1 inhibitors in the phase II trials TRUST and TRUST-II (92.5% and 92.0%),57,58,145 leading to a breakthrough therapy designation by the FDA. 154
Since the seminal approval of crizotinib, several studies have assessed the clinical activity of ROS1 inhibitors, in ROS1-TKI-pretreated patients. The ALK/ROS1 inhibitors, lorlatinib and brigatinib, were among the first to demonstrate activity in this setting with ORRs between 26.3% and 35%.135,143,155 In a small study of patients with brain-only progression while on crizotinib, lorlatinib (an agent designed specifically to penetrate the blood–brain barrier) 156 led to an ORR of 67% at 12 weeks and 87% while on the study. 137 Recently, the next-generation TRK/ROS1 inhibitors, repotrectinib and taletrectinib, have also shown promise. Repotrectinib received FDA approval in this setting based on an ORR of 38% and a mDoR of 14.8 months in the cohort of ROS1-TKI-pretreated patients from TRIDENT-1144,56,152 and taletrectinib demonstrated impressive ORRs of 52.6% and 57.1% in the cohorts of ROS1-TKI-pretreated patients from TRUST and TRUST-II.57,58,145,146 NVL-520, a ROS1-selective inhibitor designed to avoid neurotoxicity associated with TRK inhibition, showed promising clinical activity (ORR 48%) in a heavily pretreated population, including at least one prior ROS1-inhibitor, displaying both brain penetration and activity against resistance mutations. 59
In summary, both ALK/ROS1 and TRK/ROS1 inhibitors are highly active and have been approved for the treatment of NSCLC with ROS1 rearrangements; more potent and selective next-generation inhibitors continue to improve clinical efficacy and safety leading to additional regulatory approvals.
BRAF-V600-mutant
In contrast with ROS1-altered and other oncogene-driven NSCLCs (see below), high clinical activity in BRAF-V600-mutated NSCLC was only achieved thus far with a combination of both direct and downstream inhibition with BRAF60,63–65,108,157–161 and mitogen-activated protein kinase kinase (MEK)1/2 inhibitors.60,63–65,161,162 Similar to the targeting of BRAF mutations in melanoma, the use of these inhibitors as monotherapy (namely selumetinib or vemurafenib) produced minimal to modest clinical activity in NSCLC108,158,159,162 leading to combination therapy trials. The first combination to be approved was the BRAF inhibitor dabrafenib plus the MEK1/2 inhibitor trametinib for BRAF-V600E-mutated NSCLC based on outcomes from the phase II BRF113928 trial.61,62,163 At a median follow-up of 16.3–16.6 months, initial findings in both treatment-naïve and pretreated cohorts were confirmed with frequent and durable responses (ORR 63.9% and 68.4%; mDoR 10.2 and 9.8 months, respectively) and robust mPFS (10.8 and 10.2 months) and OS outcomes (17.3 and 18.2 months; Table 2). 60 The clinical activity of dabrafenib plus trametinib in this setting is also supported by an ORR of 75% from a recent phase II trial. 63
Two additional inhibitor combinations were developed to increase the potency, selectivity, and safety of at least one of the combination partners relative to prior BRAF and/or MEK inhibitors.30,164,165 The PHAROS phase II study demonstrated good activity for the second-generation combination of encorafenib plus binimetinib in treatment-naïve and pretreated cohorts (ORRs of 75% and 46%, respectively), durable responses (mDoR not yet reached (NYR) and 16.7 months), and robust mPFS (NYR and 9.3 months). 64 These findings supported approval of encorafenib plus binimetinib for the treatment of metastatic BRAF-V600E NSCLC on October 2023. 166 Recently, the HL-085-102 trial reported favorable outcomes for vemurafenib plus the highly potent and selective MEK inhibitor tunlametinib in pretreated NSCLC patients with BRAF-V600 mutations (ORR 60.6%, mDoR 11.3 months, mPFS 11.7 months). 65 Other BRAF inhibitor combinations have been tested in this setting with no breakthrough results to date.157,160,161
The development of BRAF and MEK inhibitor combinations continues to evolve with next-generation agents. A promising approach currently in early clinical testing is the use of BRAF dimer inhibitors (belvarafenib, DCC-3084, BGB-3245, PF-07799933)161,167–169 which prevent the paradoxical activation of MAPK signaling leading to resistance observed with BRAF monomer inhibitors and the need for a MEK inhibitor. These new agents are also active against Class II and III BRAF mutations.170,171
NTRK-rearranged
Inhibitors of TRK-A/B/C have been assessed in NTRK-rearranged solid tumors including the first-generation agents larotrectinib66,172 and entrectinib67,173 and the next-generation TRK inhibitors taletrectinib 146 and repotrectinib.68,174 Both larotrectinib and entrectinib were approved in 2018 and 2019, respectively, for tumor-agnostic indications in NTRK fusion-positive tumors based on integrated analyses of multicenter, single-arm trials.149,175 Up-to-date, tumor-agnostic and tumor-specific analyses have generally confirmed the benefit of these agents in lung cancer. Integrated analysis of phase I/II LOXO-TRK-14001, NAVIGATE, and SCOUT trials reported an ORR of 69% and mDoR, mPFS, and mOS of 32.9, 29.4, and NYR months for larotrectinib among 269 patients with solid tumors and an ORR of 73%, mDoR, mPFS, and mOS of 33.9, 35.4, and 40.7 months, respectively, among a subgroup of 20 lung cancer patients 66 (Table 2). Similar analyses of the phase I/II STARTRK-2, STARTRK-1, and ALKA-372-001 studies evaluating entrectinib reported an ORR of 62.4% and mDoR, mPFS, and mOS of 29.4, 15.7, and 38.2 months among 194 patients with solid tumors 173 and an ORR of 62.7% and mDoR, mPFS, and mOS of 27.3, 28.0, and 41.5 months among 51 patients with NSCLC. 67
The next-generation TRK inhibitor repotrectinib has also demonstrated clinical activity in patients with NTRK-rearranged solid tumors, 174 leading to a breakthrough therapy designation by the FDA in October 2021. 176 In a recent update of the TRIDENT-1 basket trial, repotrectinib continued to show promising clinical activity in solid tumors with ORRs of 58% and 50% in TKI-naïve and -pretreated cohorts and of 62% and 42% in the respective subsets of NSCLC patients. 68
TRK inhibitors have shown strong activity and robust clinical outcomes across different NTRK-fusion positive tumors including NSCLC, representing one of the most compelling examples of alteration-drug matching.
MET-altered
A host of agents have been assessed in MET-altered NSCLC including nonselective MET inhibitors (crizotinib48,69,140,177–182 and S49076 183 ), selective MET inhibitors (capmatinib,70,72,78,79,184,185 tepotinib,71,80,81 savolitinib,84,85,186,187 glumetinib,188,189 bozitinib, 190 ABN401, 191 SAR125844, 192 vebreltinib, 83 and gumarontinib 82 ), dual MET/X inhibitors (glesatinib, 73 BPI-9016M, 74 and OMO-1 193 ), anti-MET antibodies (onartuzumab 194 and emibetuzumab 195 ), antibody mixtures (Sym015-01 196 ), bispecific METxEGFR (amivantamab, 86 CKD-702 197 ) and METxMET (REGN5093 75 ) antibodies, and anti-MET antibody–drug conjugates (ADCs; telisotuzumab vedotin76,77,198). Multiple biomarkers and thresholds have also been used to select patients with variable clinical activity across studies with different agents and biomarker selection criteria.
MET-amplified or -overexpressed
In patients selected exclusively based on MET amplification, the multi-targeted-TKI crizotinib and the selective MET inhibitor capmatinib are the most well-studied agents and have generally shown only limited to modest clinical activity with an apparent relationship between higher MET amplification and improved outcomes in some studies.48,69,70,140,177,179,184 Other selective MET inhibitors have also shown preliminary efficacy signals in patients with MET-amplified tumors.71,184,192
Multiple MET inhibitors have been assessed in patients with MET overexpression or a variety of MET alterations. The first-in-class ADC telisotuzumab vedotin failed to meet the pre-specified response criteria for continuing enrolment in the subprotocol S1400K of Lung-MAP with an ORR of 9% 77 and displayed only modest activity in the LUMINOSITY phase II trial (ORR 22.1%). 76 Interestingly, biomarker analyses from LUMINOSITY revealed higher ORR in nonsquamous NSCLC without EGFR mutations and high MET overexpression (⩾50% by immunohistochemistry; ORR 52.2%). 76 Studies assessing small-molecule inhibitors or antibody-based agents with dual or bispecific targeting showed limited to modest activity (ORRs 2.6%–25.9%).73–75,196
METex14-mutant
Studies of MET inhibitors in patients with NSCLC harboring predominantly METex14 mutations generally resulted in more favorable clinical outcomes than those seen in MET-amplified/-overexpressed NSCLC.140,70,81,177,180,186,187 Crizotinib demonstrated variable activity in METex14 NSCLC (ORR 12%–65%),57,177,179–181 and received an FDA breakthrough therapy designation in 2018 199 based on favorable results from the PROFILE-1001 study. 182 The first regulatory approval in this setting was granted to the selective MET inhibitor capmatinib based on results from the multi-cohort GEOMETRY mono-1 phase II trial. 200 Updated results from this study showed frequent and durable responses and robust PFS and OS outcomes in both treatment-naïve (ORR 65.6%–67.9%, mDoR 12.6–NYR months, mPFS 10.8–12.4 months, mOS 20.8–NYR months in initial and expansion cohorts) and previously treated patients (ORR 40.6%–51.6%, mDoR 8.4–9.7 months, mPFS 5.4–6.9 months, mOS 13.6–NYR months in initial and expansion cohorts).70,78 More recently, the GeoMETry-III phase III trial comparing capmatinib to standard-of-care docetaxel in previously treated METex14 NSCLC showed numerical differences in ORR (53.3% vs 0%) and PFS (6.1 vs 4.1 months; hazard ratio (HR) 0.46, 95% confidence interval (CI) 0.16–1.3, p = 0.066) that although consistent with GEOMETRY mono-1 results did not reach statistical significance after early trial termination. 79 Tepotinib was the second selective MET inhibitor to receive accelerated approval for METex14 NSCLC in February 2021 based on initial results from the large phase II VISION trial. 201 Updated results confirmed initial findings with frequent and durable responses and robust PFS and OS outcomes in both treatment-naïve and pretreated patients (ORR 57.3% and 45.0%, mDoR 46.4 and 12.6 months, mPFS 12.6 and 11.0 months, mOS 21.3 and 19.3 months, respectively). 80 Additional selective MET inhibitors have also shown preliminary efficacy signals187,189–191 that were confirmed in larger cohorts of phase II studies of savolitinib, gumarontinib, and vebreltinib (ORRs 47.1%, 66%, and 75%, respectively).82–86 Notably, vebreltinib showed high activity, durable responses, and robust PFS outcomes in both treatment-naïve and pretreated cohorts of the KUNPENG study (ORR 77.1% and 70.6%; mDoR 16.5 and 15.3 months; mPFS 14.5 and 7.1 months). 83 The dual MET/OCT-2 inhibitor OMO-1 and the bispecific METxEGFR antibody CKD-702 have shown early signs of activity.193,197 The combination of capmatinib with the immune checkpoint inhibitor (ICI) spartalizumab was assessed in treatment-naïve patients resulting in a modest ORR (38.7%) and high rates of treatment-related adverse events prompting early trial termination. 185
In crizotinib-pretreated patients with predominantly skipping alterations (75%), capmatinib showed minimal activity (ORR 10%) 72 while in MET-inhibitor-pretreated patients, the bispecific METxEGFR antibody amivantamab demonstrated promising clinical activity (ORR 33%) in the METex14 cohort of the CHRYSALIS phase I trial. 86
In summary, targeting MET dysregulation with MET inhibitors has been successful in METex14 NSCLC. More potent and selective next-generation TKIs have shown high clinical activity and are the only type of agents approved thus far in this setting. In MET-amplified/-overexpressed tumors, encouraging results have been observed particularly in MET-amplified subsets. An interesting relationship between levels of MET enrichment and clinical activity was apparent in multiple studies.
RET-rearranged
Earlier generation multi-kinase inhibitors (cabozantinib, alectinib, vandetanib, lenvatinib, and ponatinib)87,202–208 and more selective RET TKIs (selpercatinib, pralsetinib, BOS172738, KL590586, SY-5007)88–95,209 have been assessed in patients with NSCLC harboring RET rearrangements. Small phase I–II trials (⩽25 patients) assessing multitargeted TKIs for which RET is a secondary target generally reported minimal activity (ORRs 4%–28%)87,202,203,205,207 except the LURET phase II trial of vandetanib which reported a promising ORR of 53%.204,208 More recently, studies of selective RET TKIs have demonstrated improved outcomes. Selpercatinib was the first agent approved by the FDA in this setting 210 based on initial results of the large, multi-cohort non-randomized LIBRETTO-001 phase II trial. 89 Trial results were recently confirmed showing frequent and durable responses and robust PFS outcomes in both treatment-naïve (ORR 84%, mDoR 20.2 months, mPFS 22.0 months) and pretreated patients (ORR 61%, mDoR 28.6 months, mPFS 24.9 months; Table 2). 88 The smaller phase II trial LIBRETTO-321 further confirmed the favorable clinical activity of selpercatinib in patients with RET-rearranged NSCLC with an ORR of 69.2% (87.5% in treatment-naïve and 61.1% in pre-treated subsets). 90 Recently, the phase III trial LIBRETTO-431 comparing selpercatinib to chemotherapy as initial treatment of RET-rearranged NSCLC met its primary endpoint with a large improvement in mPFS (24.8 vs 11.2 months; HR 0.46, p < 0.001) and more frequent and longer responses (ORR 84% vs 65%; mDoR 24.2 vs 11.5 months) for selpercatinib. 91 In September 2022, selpercatinib received full FDA approval for RET-rearranged NSCLC based on the updated results from the NSCLC cohort of the LIBRETTO-001 trial. 211 The FDA has also granted full approval to pralsetinib for the treatment of RET fusion-positive NSCLC in August 2023 212 based on the recently updated results of the phase II ARROW trial which confirmed initial findings 209 with an ORR of 72% and mDoR NYR among chemotherapy-naïve patients and ORR of 59% and mDoR of 22.3 months among platinum-pretreated patients. 92 Median PFS was NYR and 16.3 months in chemotherapy-naïve and -pretreated patients, respectively. Other highly potent and selective RET inhibitors (BOS172738, KL590586, and SY-5007) have shown promising clinical activity in phase I trials (ORR 33%–76%).93–95 Similar to other alteration-matched settings, selective RET TKIs have improved outcomes relative to multitargeted TKIs in the treatment of RET-arranged NSCLC; selpercatinib and pralsetinib are the only agents approved in this setting.
HER2-altered
Four main types of inhibitors, dual- or pan-HER TKIs (BAY2927088, neratinib, dacomitinib, afatinib, pyrotinib, poziotinib, and tarloxotinib-effector),96–103,213–221 selective HER2 TKIs (zongertinib),104,105,222 anti-HER2 monoclonal antibodies (mAbs; trastuzumab, pertuzumab, and inetetamab),106–109,223–227 and anti-HER2 ADCs (ado-trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-DXd))110–115,228–232 have been assessed in patients with NSCLC with HER2 alterations.
Second-generation small-molecule TKIs that were primarily developed as EGFR inhibitors in NSCLC (dacomitinib, afatinib, and neratinib) were tested in HER2-altered NSCLC showing limited activity as single agents (ORRs 0%–12%).213,215,216,218,219 More recently, newer next-generation multi-HER TKIs (poziotinib, pyrotinib, tarloxotinib, BAY2927088, BDTX-189) and HER2-selective TKIs (zongertinib) have shown better yet generally modest activity in primarily HER2ex20-mutated NSCLC (ORR 19.2%–60%; Table 2).96–105,214,220–222 Of these, the highest ORR was observed with the multi-HER TKI BAY2927088 (60%) 103 leading to an FDA breakthrough designation for HER2-mutated NSCLC. 223 No small-molecule HER2 TKI has been approved to date for the treatment of HER2-altered NSCLC.
Antibody-based agents have the potential to increase selectivity and specificity relative to multitargeted TKIs. However, the use of single or dual anti-HER2 mAb regimens with or without chemotherapy has resulted in minimal to modest activity in HER2-altered NSCLC (0%–45%; Table 2).106–109,224–228 Recently, two ADCs, T-DM1 and T-DXd, have been prospectively assessed in this setting. T-DM1, which delivers a microtubule-inhibitory payload to HER2-presenting cells, has shown minimal activity in patients with pretreated HER2-amplified/-overexpressed NSCLC (ORR 6.7%–20%)229–231 and moderate activity in chemotherapy-pretreated patients with HER2ex20 insertions (ORR 38.1%). 110 T-DM1 also showed limited activity when combined with osimertinib in osimertinib-pretreated patients with HER2 overexpression. 111 T-DXd, which delivers a topoisomerase 1 inhibitor payload, has shown greater activity overall in HER2-altered (including overexpressed and mutated) NSCLC with ORRs of 55.6% and 55% in heavily pretreated patients from a phase I study dose-expansion cohort and the phase II DESTINY-Lung01 trial, respectively.113,114,232 Both studies reported high ORRs in patients with HER2 mutations compared with HER2 overexpression (72.7% and 61.9% vs 26.5%–34.1%, respectively).112,113,232 Longer mDoR, mPFS, and mOS in HER2-mutated relative to HER2-overexpressed patients were also apparent in DESTINY-Lung01 (NYR vs 5.8–6.2 months, 14.0 vs 5.7–6.7 months, and NYR vs 11.2–12.4 months, respectively). T-DXd received a breakthrough therapy designation for use in platinum-pretreated HER2-mutant NSCLC patients from the FDA in May 2020 based on results of DESTINY-Lung01. 233 More recently, results from the randomized phase II trial DESTINY-Lung2 comparing two doses of T-DXd (5.4 vs 6.4 mg/kg every 3 weeks (q3w)), confirmed the favorable outcomes (ORR 49.0% and 56.0%; mDoR 16.8 months and NYR; mPFS 9.9 and 15.4 months; mOS 19.5 months and NYR for the 5.4 and 6.4 mg/kg q3w doses, respectively) 115 leading to the FDA granting only the lower-dose regimen accelerated approval for use in HER2-mutant NSCLC in August 2022 due to concerns of higher rates of interstitial lung disease/pneumonitis with 6.4 mg/kg q3w. 234 This regimen is also being evaluated for a HER2-amplified, tumor-agnostic indication with promising efficacy. 235 Other HER2-directed ADCs (A166 and ARX788) and bispecific antibodies (KN026) have shown promising results in early-phase studies in HER2-altered tumors; however, their efficacy in NSCLC has yet to be established.236–238
Several clinical studies now show a greater benefit for HER2-directed therapy in those with HER2 mutations compared to other types of HER2 alterations, with the best outcomes observed for the ADC T-DXd which was approved for HER2-mutated NSCLC.
KRAS-mutant
Multiple approaches have sought to target KRAS mutations in NSCLC, including direct inhibition with RAS/RAF blockers125,239,240 and indirect approaches such as inhibition of downstream effectors,125,126,240–251 cyclin-dependent kinases 4/6,252–254 and other targets.125,242,247,249,250,255 The first successful effort to directly target KRAS in NSCLC emerged from the development of agents that selectively and irreversibly bind and stabilize the KRAS-G12C inactive form (single-OFF KRAS-G12C inhibitors).118,256–259 Sotorasib and adagrasib received accelerated approval from the FDA in May 2021 and December 2022260,261 for previously treated, KRAS-G12C-mutant NSCLC based on results from the multicenter, single-arm trials CodeBreaK100 and KRYSTAL-1, respectively.118,258 Initial results from CodeBreaK100 were confirmed in a 2-year update showing frequent and durable responses and robust time-to-event outcomes (ORR of 41% and mDoR, mPFS, and mOS of 12.3, 6.3, and 12.5 months, respectively). 116 After a median follow-up of 17.7 months, the confirmatory, phase III trial CodeBreaK200 met its primary endpoint of PFS (median 5.6 vs 4.5 months, HR 0.66, p = 0.0017) and showed significant improvement in ORR (28.1% vs 13.2%, p < 0.001) with sotorasib relative to docetaxel in platinum-pretreated patients. 117 However, benefits in mDoR (8.6 months (95% CI 7.1–18.0) vs 6.8 months (95% CI 4.3–8.3)) and OS (10.6 vs 11.3 months, p = 0.53) were not apparent which may be in part due to removal of OS as a co-primary endpoint, consequent reduction in sample size and introduction of crossover from docetaxel to sotorasib in a trial amendment. 262 Despite CodeBreaK200 meeting its primary endpoint, the FDA ruled that the primary endpoint data could not be reliably interpreted and postponed conversion to full approval. 263 Initial results from the KRYSTAL-1 trial leading to accelerated approval of adagrasib were comparable to sotorasib’s registrational data with an ORR of 42.9% and mDoR, mPFS, and mOS of 8.5, 6.5, and 12.6 months, respectively. 118 Adagrasib’s confirmatory trial KRYSTAL-12 is ongoing. More recently, the new potent and selective G12C inhibitors divarasib, JDQ443, and garsorasib have shown promising activity as single agents with ORRs of 53.4%, 41.7%, and 38.7%, mDoR of 14.0, not reported (NR) and 6.9 months, and mPFS of 13.1, NR and 7.6 months, respectively.119–121 Additional single-OFF G12C inhibitors are either in very early stages of clinical development or have been halted due to safety and/or efficacy concerns.264–268
New RAS inhibitors are now being developed toward different individual mutations (KRAS-G12X) or with a wider selectivity range (from multi-KRAS to multi-RAS), and targeting active (ON) forms. 259 Recently, the first-in-class, RAS-selective, tri-complex multi-ON RAS inhibitor, RMC-6236, showed promising preliminary clinical activity in KRAS-G12X(D/V/A/S/R) NSCLC (ORR 38%; Table 2). 129
Data on combination regimens in patients with KRAS-G12C mutations have recently emerged. Combination of sotorasib with ICIs (atezolizumab or pembrolizumab) showed only moderate activity (ORR 29%) in cohorts of the CodeBreak100/101 trials where frequent grade 3/4 hepatotoxicity limited ability to maintain dosing. 122 Higher ORRs were observed with the selective KRAS-G12C inhibitors MK-1084 and adagrasib in combination with pembrolizumab (47% and 63%, respectively) in programmed death-ligand 1 (PD-L1)-positive (tumor proportion score (TPS) ⩾1%) and PD-L1-high (TPS ⩾ 50%) patients, respectively.127,269 Combinations of sotorasib or glecirasib with SHP2 inhibitors (RMC4630 or JAB-3312, respectively) in pretreated patients showed moderate ORRs overall (43% and 27%, respectively) with promising activity in G12C inhibitor-naïve subsets (ORR of 50% in both studies).123,124 Combinations of MEK inhibitors (binimetinib, trametinib, or avutometinib) with other systemic agents (chemotherapy, defactinib, or multi-TKIs (erlotinib, anlotinib, ponatinib)) have shown variable activity in pretreated patients with KRAS mutations (ORRs 0%–60%).125,126,242,243,246–249 In chemotherapy-naïve non-squamous NSCLC patients, first-line sotorasib plus carboplatin-pemetrexed showed an impressive ORR of 88.9% and PFS and OS rate at 6 months of 81.2% and 87.0% in the SCARLET phase II trial. 128
In summary, the development of agents that indirectly target KRAS (such as MEK/ERK inhibitors) has not been successful in KRAS-mutant NSCLC. KRAS-G12C single-OFF inhibitors have shown strong activity in previously treated, KRAS-G12C-mutant NSCLC and are the only type of targeted agents approved in this setting. New single- and multi-ON inhibitors are promising agents as they can more directly inhibit active RAS forms and have the potential to simultaneously inhibit different aberrant forms; however, these are still in very early stages of development.
Other targets
Several targeted therapies are being developed in alteration-drug-matched settings without a first-in-class regulatory approval to date, including FGFR-, HER3-, NRG1-, PTK7-, and PI3K-altered NSCLC.
FGFR inhibition of FGFR-altered NSCLC was initially attempted with non-selective inhibitors mostly in FGFR-amplified/overexpressed tumors resulting in considerable toxicity with minimal activity.270–273 The use of FGFR-selective, multi- or pan-FGFR inhibitors eased safety concerns; however, clinical activity remained minimal (ORRs 4%–11%).274–277 The recently presented results of the FIND and RAGNAR trials of erdafitinib followed this trend with minimal to modest activity in NSCLC patients with FGFR alterations (ORRs of 9% and 26%, respectively).130,278 Moreover, rogaratinib treatment of squamous NSCLC with FGFR alterations produced no responses in SAKK19/18 which was closed prematurely due to futility. 279
In addition to inhibition of HER2-altered NSCLC, multiple approaches have sought to address aberrant HER signaling, including anti-HER3 mAbs and small-molecule pan-HER inhibitors in patients with HER3 or NRG1 overexpression/amplification or NRG1 rearrangements. Testing of small-molecule pan-HER inhibitors in HER1-3-altered NSCLC had limited success.280–282 Although circulating NRG1 levels were initially identified as potentially predictive of efficacy of the anti-HER3 mAb patritumab plus erlotinib in the randomized phase II HERALD trial, 283 the phase III HER3-Lung study failed to confirm this finding. 284 Similarly, the addition of the anti-HER3 mAbs lumretuzumab or seribantumab to either chemotherapy or EGFR inhibitors did not show meaningful benefit (ORRs of 6.3% and 19.7% in pretreated patients and 42.9% in a small subset of chemotherapy-naïve patients) despite early signals of higher activity in HER3- or NRG1-enriched NSCLC.285–288 The anti-HER3 mAb GSK2849330 showed minimal activity in HER3-expressing cancers (n = 29) with a single yet durable response in an NSCLC patient with a cluster of differentiation 74-NRG1 rearrangement. 289 When NRG1 rearrangements were used as biomarker selection criteria, seribantumab and the HER2/HER3 bispecific antibody zenocutuzumab showed moderate yet promising activity with durable responses in previously treated NSCLC patients enrolled in the CRESTONE (ORR 36%) 290 and eNRGy trials (ORR 37.2%; mDoR 14.9 months). 131 These agents have received fast-track or priority review designations from the FDA in NRG1-rearranged tumor-agnostic or NSCLC-specific indications.291,292
Development of therapies targeted to PI3K and PTK7 in the respective biomarker alteration-matched NSCLC populations is in the very early stages of development without a clear breakthrough to date.132,293,294
Discussion
Development of therapies directed toward driver genes in molecularly selected, advanced NSCLC is an extremely active research field with a large number of studies evaluating new agents in previously identified targets (particularly in MET-, HER2-, and KRAS-altered disease) and new potentially actionable molecular targets (NRG1 and PTK7). MET, HER2, KRAS, ROS1, RET, and BRAF alterations are both clinically actionable and relatively common in NSCLC patients (>1%). It is therefore not surprising that these are among the most studied populations. Although NTRK alterations are uncommon (⩽1%), their clinical actionability has been convincingly demonstrated independent of tumor type, with high clinical activity across multiple tumors and several tumor-agnostic approvals.
The number of unique clinically actionable settings defined by oncogenic alterations (other than those of EGFR and ALK) continues to grow and clinical research efforts in this field have led to a large number of FDA approvals in the last 3 years (n = 9). 295 Many of these are in patient populations with a high clinical need as oncogenic alterations tend toward more aggressive cancers with few treatment options. The clinical impact of alteration-matched therapies is also reflected in trial eligibility for new non-oncogenic-targeted agents (such as ICIs) which now often exclude patients with clinically actionable alterations (EGFR, ALK, ROS1, and others). Moreover, approval of tumor-agnostic indications for the same alteration-drug pairs previously approved in NSCLC296,297 serves as further validation of the applicability of this strategy. However, despite efforts to standardize reporting and interpretation of alteration-drug-matched clinical trial data,298–302 regulatory approvals and reimbursement vary across regions limiting access in certain jurisdictions.303–308
Our review highlights the considerable development of targeted therapy in NSCLC, resulting in an overall increase in the antitumor activity of alteration-drug-matched strategies across multiple oncogene-driven settings (Figure 2). Fueled by developments in molecular diagnostic tools, patient selection has evolved from biomarker-unselected populations to those defined by an altered biomarker and, more recently, by specific alterations with known oncogenic potential. Target actionability has also improved through the development of increasingly selective and potent agents, with better pharmacokinetic profiles, that are capable of inhibiting specific alterations at lower doses and with fewer off-target effects. Although the initial use of multi-targeted TKIs allowed multiple alterations to become simultaneously actionable (e.g., crizotinib in ALK-, ROS1-, and MET-altered disease), these agents have generally been replaced with more potent, selective, and/or direct small-molecule inhibitors or antibody-based agents. For example, even though crizotinib had initially shown considerable activity in ROS1-rearranged NSCLC, ORRs in this setting continue to improve with next-generation inhibitors that display higher selectivity toward ROS1 and its mutant forms (Figure 2).57–59,309 Nonetheless, strategies involving simultaneous inhibition of multiple targets such as co- and pan-inhibitory approaches may still be useful in areas where a more stringent biological control is required due to compensatory mechanisms (such as functional redundancy and pathway feedback loops) or weak primary target inhibition. With the establishment of alteration-drug-matched approaches as first-line therapy in the advanced setting, there is an increasing need for strategies to overcome both innate and acquired resistance. 310 These commonly involve co-inhibition with combination therapy or bispecific agents to address off-target mechanisms (e.g., KRAS-G12C plus SHP2 inhibitors against adaptative resistance to KRAS inhibition123,124,310 or amivantamab to overcome reciprocal resistance and signaling crosstalk between EGFR and MET)310–312 and/or use of next-generation inhibitors which typically address on-target resistance mechanisms (e.g., taletrectinib against the ROS1 secondary solvent-front mutation G2032R). 309 Current research also increasingly favors direct alteration targeting over indirect strategies such as modulation of proximal “pathway” components or levels of effector molecules or by stabilizing inactive/OFF states. Newer antibody-based agents are improving the clinical actionability of alteration-matched approaches by directed delivery of cytotoxic moieties (ADCs) or by specifically and simultaneously targeting multiple alterations with potential synergistic effects (bispecific antibodies). Bispecific antibodies have also the potential of combining alteration-drug-matching with other therapeutic approaches that have been successful in the treatment of NSCLC, such as immune modulation of the tumor microenvironment with ICIs.

Clinical activity of selected types of agents used as initial targeted therapy across different oncogene-driven NSCLC settings.
Development of effective therapeutic strategies has been challenging in some biomarker-selected settings, such as PI3K- and FGFR-altered NSCLC for which no agent was approved despite decades-long research efforts. In particular, alterations to PI3K and its efferent (PI3K-AKT-mammalian target of rapamycin (mTOR)) pathway are relatively common in NSCLC and multiple direct and indirect inhibitory approaches have been attempted with pan-class I PI3K, PI3K subtype, AKT, mTOR, and PI3K/mTOR inhibitors in both alteration-drug-matched and -unmatched populations.316–318 PI3K-AKT-mTOR is an example of a pathway with complex regulatory mechanisms and intricate crosstalk, where target inhibition is challenged by multiple intra- and inter-pathway compensatory mechanisms (including functional redundancy of PI3K isoforms) and on-target toxicities.316–320 Moreover, PI3K pathway alterations are genetically diverse, occur in a clinically heterogenous group of patients, and are often associated with alterations in other oncogenes and high mutational load, where they may be “passengers” rather than “drivers” of the oncogenic process.320–322 In addition to addressing issues with inhibitor selectivity and toxicity, and similar to MET inhibitors in MET-altered disease, a critical step in the development of PI3K inhibitors in NSCLC may be the identification and targeting of oncogenic drivers among the range of PI3K pathway alterations. 320
It is important to note that many targeted agents are currently being developed in unselected/non-matched populations, using indirect (“pathway”) inhibition strategies and targeting non-oncogene tumor-associated alterations. For example, treatment of NFE2L2/KEAP1-altered NSCLC is being attempted indirectly with glutaminase 323 and mTOR inhibitors.324,325 ADCs targeting the human trophoblast cell surface glycoprotein antigen 2 (TROP2; datopotamab deruxtecan, sacituzumab govitecan, and SKB264) have recently shown promise in NSCLC in combination with ICIs326,327 or as single agents.328–330 However, their development has been directed toward TROP2-unselected populations as biomarker analyses have failed to establish TROP2 expression as a predictor of clinical activity.328,331 Development of ADCs against other tumor-associated markers especially in populations with high levels of target expression is a promising approach that is currently being explored.332–334
Summary
Research in alteration-drug matching continues to evolve at a rapid pace in NSCLC. The number of settings defined by oncogenic alterations has increased and alteration targeting has become increasingly specific and effective with the refinement of biomarker selection criteria and the use of newer, more selective, and potent agents. Future developments should focus on the continued application of these principles to new settings and the exploration of novel ways to target oncogene-driven NSCLC.
Supplemental Material
sj-docx-1-tam-10.1177_17588359241308784 – Supplemental material for The continually evolving landscape of novel therapies in oncogene-driven advanced non-small-cell lung cancer
Supplemental material, sj-docx-1-tam-10.1177_17588359241308784 for The continually evolving landscape of novel therapies in oncogene-driven advanced non-small-cell lung cancer by Barbara Melosky, Rosalyn A. Juergens, Shantanu Banerji, Adrian Sacher, Paul Wheatley-Price, Stephanie Snow, Ming-Sound Tsao, Natasha B. Leighl, Ilidio Martins, Parneet Cheema, Geoffrey Liu and Quincy S. C. Chu in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.