
Abstract
Background:
Cholangiocarcinoma is a kind of malignant tumor that originates in the epithelium of the biliary tract. Although there are several options for second-line treatment for patients without specific genetic mutations, the overall treatment efficacy is disappointing. Second-line treatment which is composed of liposomal irinotecan plus fluorouracil and leucovorin significantly improved the treatment efficacy for advanced biliary tract cancer and extended patient survival. This study aims to evaluate the efficacy and safety of the combination of cadonilimab with liposomal irinotecan plus fluorouracil and leucovorin for advanced biliary tract cancer.
Objectives:
The primary objective of this study is to determine the objective response rate. The second objectives of this study are overall survival, progression-free survival, disease control rate, and adverse event incidence rate.
Design:
The study is a single-arm, prospective phase II clinical trial. In all, 51 patients who are diagnosed with locally advanced or metastatic bile tract cancer will be enrolled.
Methods and analysis:
Eligible participants will receive cadonilimab at a dosage of 6 mg/kg on day 1 of each 14-day cycle combined with intravenous liposomal irinotecan at a dosage of 70 mg/m2 for 90 min on day 1 plus leucovorin at a dosage of 400 mg/m2 for 30 min on day 1 and fluorouracil at a dosage of 2400 mg/m2 for 46 h every 2 weeks.
Discussion:
Previous studies have suggested that there is a synergistic effect between the two treatment modalities. However, the potential of cadonilimab in bile tract cancer has not been explored. Hence, this trial is the first to investigate its efficacy and toxicity. In addition, the trial is also willing to explore potential biomarkers in patients with locally advanced and metastatic bile tract cancer.
Trial registration:
This study was registered on ClinicalTrials.gov with NCT06438822.
Ethics:
This study protocol and amendments have been approved by the Ethics Committee of West China Hospital (2024(791)).
Introduction
Cholangiocarcinoma, also known as bile tract cancer, is one of the malignant tumors originating from the epithelium of the biliary tract and can be anatomically classified as intrahepatic, hilar, extrahepatic, or gallbladder cholangiocarcinoma. 1 Due to the extremely invasive nature of cholangiocarcinoma, most patients have missed their chance at curative surgery at the time of diagnosis. In addition, cholangiocarcinoma exhibits limited responsiveness to chemotherapy, leading to a bleak prognosis, with an overall 5-year survival rate of fewer than 10%. 2
Chemotherapy dominates the treatment modality for locally advanced unresectable or metastatic cholangiocarcinoma. The ABC-02 research confirmed that the GemCis regimen which consisted of gemcitabine plus cisplatin was effective as the initial treatment for cholangiocarcinoma. 3 Recently, the advancement of immunotherapy has led to notable breakthroughs in the treatment of cholangiocarcinoma with the combination of immune checkpoint inhibitors and chemotherapy.
Patients who have shown disease progression after first-line treatment have various alternatives for second-line treatment, although the overall effectiveness is unsatisfactory. The ABC-06 study 4 was the sole extensive randomized phase III trial conducted to evaluate second-line treatment, and it suggested that chemotherapy with the FOLFOX regimen, which was composed of oxaliplatin, folinate acid, and fluorouracil, in combination with best supportive care could improve patient survival. Although the results of this study have statistical significance, its actual efficacy and tolerability may not be satisfactory to clinicians.
Liposomal irinotecan is a novel antitumor drug that utilizes liposomal technology to encapsulate irinotecan within a double layer of phospholipids. Two clinical trials (NAPOLI-1 and NAPOLI-3)5,6 have confirmed the effectiveness of liposomal irinotecan as a first-line and second-line treatment for pancreatic cancer and have been recommended in guidelines. In advanced biliary tract cancer, the NIFTY study7,8 revealed that the second-line NIFU regimen of liposomal irinotecan plus fluorouracil and leucovorin greatly enhanced the effectiveness of therapy for advanced cholangiocarcinoma and extended the duration of survival.
Cadonilimab is a novel immune checkpoint inhibitor belonging to the programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) dual-specific antibody targeting class that was developed independently in China. 9 This symmetric tetravalent bispecific antibody is designed to bind with high affinity to PD-1 and CTLA-4, both co-expressed on tumor-infiltrating lymphocytes, ensuring efficient co-targeting. A phase Ib/II clinical studies have suggested that cadonilimab has remarkable efficacy in treating various malignancies, including cervical cancer and liver cancer. 10 In the phase Ib dose-escalation stage, no dose-limiting toxicities were observed, and cadonilimab demonstrated an overall favorable safety profile. The incidence of Grade 3–4 treatment-related adverse events (TRAEs) was 27.9%. The most common Grade ⩾3 TRAEs included anemia, decreased appetite, neutropenia, and infusion-related reactions. In a clinical trial investigating the use of cadonilimab as monotherapy for advanced solid tumors, six advanced colorectal cancer patients with microsatellite instability-high (MSI-H) or deficient mismatch repair (dMMR) were enrolled. All six patients achieved partial response (PR), resulting in an objective response rate (ORR) of 100%. 11 It has received approval as a standalone treatment for advanced cervical cancer in the second-line setting.
Hence, we hypothesize that the utilization of the NIFU regimen in conjunction with cadonilimab has the potential to augment effectiveness in the treatment of biliary tract cancer in the second-line setting.12,13 The objective of this trial is to assess the effectiveness and safety of combining cadonilimab with the NIFU regimen for treating advanced biliary tract cancer that has not responded to the first gemcitabine chemotherapy. Furthermore, our objective is to investigate pertinent predictive biomarkers that can accurately determine the effectiveness of treatments, to offer significant knowledge for use in clinical practice.
Methods
Study design
The study is a prospective, single-arm, exploratory phase II clinical trial conducted at the West China Hospital of Sichuan University, Peking University Cancer Hospital, etc. This study protocol and amendments have been approved by the Ethics Committee of West China Hospital (2024(791)). And it was registered on ClinicalTrials.gov with NCT06438822. This study aims to assess the effectiveness and safety of combining cadonilimab with the NIFU regimen for treating patients with advanced biliary tract cancer that has not responded to gemcitabine chemotherapy as first-line treatment (Figure 1). Written informed consent forms will be required from all study participants prior to any study-specific procedures being conducted. A total of 51 patients will receive cadonilimab combined with the NIFU regimen, which is composed of intravenous liposomal irinotecan (70 mg/m2 for 90 min) plus intravenous leucovorin (400 mg/m2 for 30 min) and intravenous fluorouracil (2400 mg/m2 for 46 h), every 2 weeks. There will be two cohorts. Cohort 1 consists of patients who experienced treatment failure in the initial line of therapy using gemcitabine-based chemotherapy in combination with programmed cell death ligand 1 (PD-L1)/PD-1 monoclonal antibody immunotherapy. Cohort 2 comprises patients who have experienced treatment failure with gemcitabine-based chemotherapy as their initial treatment and have not undergone PD-L1/PD-1 monoclonal antibody immunotherapy. Upon recruitment, participants must adhere to the trial protocol until there is an imaging-confirmed progressive illness, fatality, severe adverse events (AEs), or when patient or investigator needs are fulfilled. This protocol completely adheres to the Standard Protocol Items: Recommendations for Interventional Trials statement. 14

The main process of this clinical trial.
Study endpoints
Primary endpoint
The primary objective of this study is to determine the ORR. The validity of the total number of individuals in the intention-to-treat (ITT) population who achieve a complete response (CR) or PR will be assessed. The statistical results obtained from imaging (Computed Tomography [CT] or Magnetic Resonance Imaging [MRI]) using the RECIST v1.1 criteria will be utilized to compute the ORR.
Secondary endpoints
The disease control rate (DCR) is calculated as the proportion of people in the ITT population whose tumor response is classified as CR, PR, or stable disease (SD), based on CT/MRI scans using the RECIST v1.1 criteria.
Progression-free survival (PFS) is the length of time from the initial administration of treatment to the occurrence of imaging-confirmed PD, severe AEs, death, or the last evaluation of tumor response.
Overall survival (OS) refers to the period of time starting from the first administration of medical treatment until either death or loss of follow-up occurs.
AE incidence rate is the ratio of the number of patients experiencing AEs to the total number of patients included in the evaluation of AEs.
Study population
Inclusion and exclusion criteria
The patients who comply with the inclusion criteria are advanced biliary tract cancer in which primary chemotherapy with gemcitabine failed. In addition, patients with an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–1, sufficient organ function, and no history of active autoimmune disease are eligible for this trial (essential inclusion and exclusion criteria are elaborated in Table 1).
The key eligible criteria of this trial.

ALT, alanine aminotransferase; AST, aspartate aminotransferase; ECOG PS, Eastern Cooperative Oncology Group performance status; ULN, upper limit of normal.
Baseline evaluation
Following the screening process, a baseline assessment will be conducted within 2 weeks prior to the commencement of medication. Eligible patients will undergo a comprehensive data collection procedure, including written informed consent, medical history, comprehensive physical examination results (including vital signs), ECOG PS, laboratory test results, information about tumors (including CT or MRI, tumor markers, diagnoses, and stages of primary tumor), and demographic details.
Intervention
Eligible participants will receive cadonilimab at a dose of 6 mg/kg on the first day of a 14-day cycle combined with intravenous liposomal irinotecan at a dose of 70 mg/m2 for 90 min on the first day, along with leucovorin at a dose of 400 mg/m2 for 30 min on the first day and fluorouracil at a dose of 2400 mg/m2 for 46 h every 2 weeks.
Unless the subject progresses or has an intolerable toxic reaction, CT or MRI scans will be conducted every two cycles to assess the tumor response based on RECIST version 1.1 and immune-related RECIST (ir RECIST). Patients with SD or a partial or CR will continue treatment. Participants will receive maintenance treatment with cadonilimab for a maximum of 24 months at the end of 12 cycles of NIFU regimen chemotherapy combined with cadonilimab. PD will result in withdrawal of the trial, and if the participant’s efficacy evaluation has not progressed by the end of the therapy phase of the study, the participant may continue to be treated according to the protocol, and the investigator will continue to follow the subject for the duration of the treatment period until the disease has progressed or until the subject begins a new antitumor therapy.
Assessment and follow-up
Prior to each cycle, a comprehensive assessment will be implemented to ensure that the subject is suitable for the next administration. It is essential for subjects to possess adequate bone marrow function before each administration, and nonhematological toxicity not exceeding Grade 1 according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 5.0.
The evaluation of tumor response will be conducted using CT or MRI imaging techniques, in accordance with RECIST 1.1, every 8 weeks after the commencement of treatment. CT or MRI is used at the discretion of the investigator, but the method of evaluation and the machine and technical parameters should be as consistent as possible throughout the study period. Pseudoprogression may occur with treatment with cadonilimab, either due to immune cell infiltration or other mechanisms resulting in a substantial rise in preexisting tumor lesions or the emergence of new tumor lesions. Therefore, if the investigator suspects that disease progression is pseudoprogression, confirmation of disease progression by reimaging will be required after at least 4 weeks or at the time of the next scheduled imaging evaluation (it should be noted that reimaging must be done within 9 weeks after the initial confirmation of disease progression).
At the end of the trial (28 ± 7 days after stopping the drug or before starting a new antitumor therapy), a thorough assessment will be performed to evaluate the patient’s overall condition by physical examination, ECOG score, and CT or MRI scans. AEs and concomitant medications are recorded. For subjects who discontinue participation or withdraw from the study, record the reason for treatment interruption, the date of discontinuation, the date and reason for withdrawal, and the cause of death if it occurs will be recorded. If a subject is unable to proceed with the investigation due to factors unrelated to disease progression, for example, AEs, violation of the trial protocol, withdrawal of informed consent, self-withdrawal, etc., it is necessary to continue to follow the patient thereafter until the patient progresses or dies.
Subjects will be contacted by telephone at 12-week intervals starting at the end of treatment to assess survival until the subject dies or the study is terminated. Survival, subsequent antitumor therapy, and drug-related serious adverse event (SAE) data will be collected during these visits.
Safety assessment
AEs will be assessed based on pretreatment general condition data collected in accordance with the NCI-CTCAE version 5.0. Furthermore, the correlation between AEs and the research agents will be assessed to identify TRAEs. In the event of disability, life-threatening illness, fatal, hospitalization or prolonged hospitalization, continued or severe disability/dysfunction, AEs will be classified as SAEs. All AEs will be documented and reported in the case report form.
If a participant experiences toxicities of varying severity, dose adjustments should be based on the highest observed toxicity level. Prior to each administration, all toxicities must be resolved to CTCAE Grade 0–1 or baseline levels, except for alopecia, fatigue, Grade 2 toxicities deemed tolerable by the participant, or other situations deemed clinically insignificant by the investigator. If a participant cannot meet the dosing criteria within the scheduled interval due to toxicities, the current cycle’s dosing will be delayed, and the timing for the next cycle will be calculated based on the actual dosing time of the current cycle. Efforts should be made to synchronize the dosing schedule of cadonilimab with that of chemotherapy drugs whenever possible.
If the investigator determines that the toxicity is primarily related to a specific drug, dose adjustments for other drugs are not required. All dose adjustments should be documented, including the rationale and actions taken.
Modification of allocated intervention
Suspension, adjustment, and delay of cadonilimab
There were no allowed modifications to the dosage of cadonilimab during the study, and temporary interruptions of up to 12 weeks were avoided. In the event of drug-related toxicity according to NCI-CTCAE version 5.0, it is recommended that the dose of cadonilimab be adjusted according to Table 2.
Recommended dose adjustment regimen for cadonilimab.

The study medication is administered when the dosage of corticosteroids is gradually reduced and the level of toxicity reverts to Grade 0–1. If the side effects of the medication do not improve to Grade 0–1 after 12 weeks of stopping the treatment, or if the dose of corticosteroids cannot be lowered to 10 mg prednisone per day or less within 12 weeks, the drug should be stopped permanently.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BV, baseline value; DRESS, drug rash with eosinophilia and systemic symptoms; GBS, Guillain–Barré syndrome; SJS, Stevens–Johnson syndrome; TEN, toxic epidermal necrolysis; ULN, upper limit of normal.
When hepatotoxicity occurs, initial management usually involves liver protection. If there is insufficient improvement after 3 days, consideration should be given to immune-related hepatitis, and treatment with corticosteroids may be warranted.
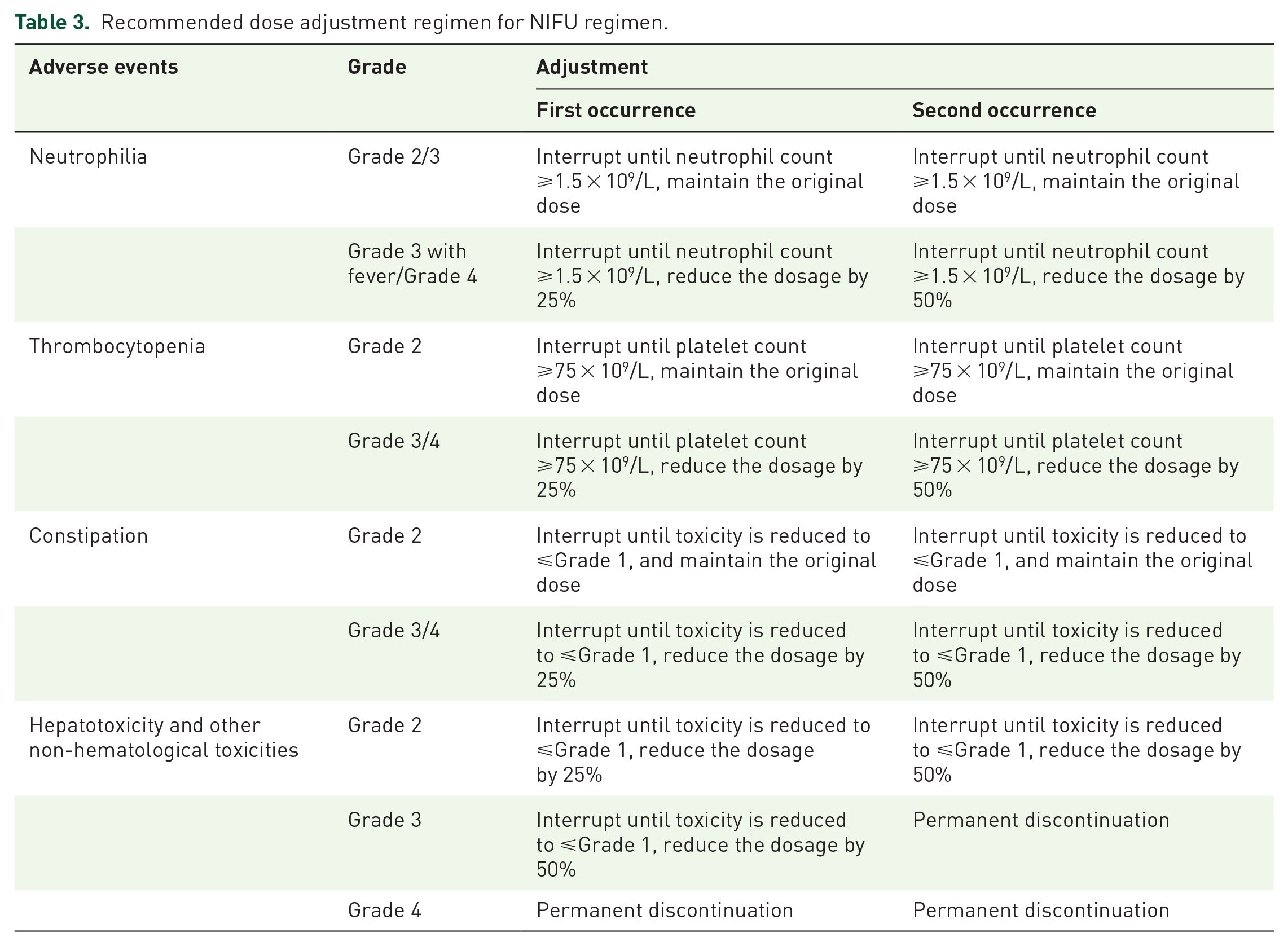
Suspension, adjustment, and delay of the NIFU regimen
Treatment suspension or dose reduction can be implemented at any point during the study to effectively manage toxic effects according to NCI-CTCAE version 5.0. It is recommended that the dose of the NIFU regimen be adjusted according to Table 3 and the general principles are as follows:
For adverse reactions, appropriate treatment should be used to improve the patient’s signs and symptoms, including the administration of antidiarrheal agents for diarrhea, antiemetic drugs to address nausea and vomiting, hepatoprotective drugs to improve abnormalities in liver function, and granulocyte colony-stimulating factor to boost leukocyte production.
Any preexisting AEs at baseline, if deemed necessary by the investigator, dose adjustments will be made based on changes in toxicity grading.
If multiple adverse reactions are concurrently present, then the most conservative dose adjustment regimen should be used.
The dose that was reduced in the event of an adverse reaction should not be increased again, and the dose should not be reduced more than twice per patient.
At the beginning of each cycle, patients may begin a new treatment cycle if their hematologic toxicity recovers to ⩽Grade 1. Otherwise, treatment may be delayed for up to 2 weeks pending the recovery of hematologic parameters. If recovery has not occurred after a 2-week delay (i.e., 4 weeks since the last chemotherapy administration), patients should permanently discontinue liposomal irinotecan.
If the investigator determines that the hazardous response is unlikely to progress into a severe or life-threatening occurrence (e.g., alopecia), the patient may continue treatment at the original dose without having to reduce or interrupt the dose. Nonhemolytic anemia may not require dose reduction or interruption of therapy if it can be improved by blood transfusion, erythropoietin, or iron therapy.
Recommended dose adjustment regimen for NIFU regimen.
Statistical design
Sample size
Based on the ORR of 5% observed in the ABC-06 clinical trial, this study is expected to increase the ORR of Cohort 1 to 20%. With a power of 80% and a type I error (one-sided) of 0.05, the sample size is conducted using two-stage phase II clinical trials with PASS 2021 software. Cohort 1 will consist of a total of 32 patients that will be enrolled, accounting for a 10% drop-out rate. Phase I will consist of 10 patients, and if none of the patients have the best response of CR or PR, the trial will be halted. Subsequently, phase II will enroll 19 patients. If at least four patients achieved the best response of CR or PR, Cohort 1 would reach the primary endpoint. Moreover, this study longings to increase the ORR of Cohort 2 to 25%. With a power of 80% and a type I error (one-sided) of 0.05, a total of 19 subjects will be enrolled in Cohort 2, accounting for a 10% drop-out rate. Phase I will consist of nine patients; if a total of greater than or equal to one patient achieves the best response of CR or PR, the trial will continue to phase II. Afterward, eight patients will be enrolled. If a total of greater than or equal to three patients had the best response of CR or PR, Cohort 2 would reach the primary endpoint. There were 51 patients in Cohorts 1 and 2.
Data analysis
The statistical analysis will be conducted using SPSS 27.0 (IBM Corporation, Armonk, NY, USA) or a more advanced version. A p value below 0.05 was deemed statistically significant for all statistical tests. The data are showcased as the mean ± standard deviation (SD) or median (range), while the count data are presented as the frequency and percentage.
This study will use the principle of ITT, which means that all subjects who provide informed consent will be analyzed. Three populations have been established to assess treatment efficacy and safety. Safety analysis will use a safety analysis set (SAS). Efficacy analysis will use a full analysis set (FAS) and per protocol set (PPS).
The FAS represents the dataset obtained from all subjects in which the study medication was used at least once in accordance with the protocol.
PPS is a subset of FAS that is defined as individuals who have received study medication and did not experience a major violation of the study protocol.
SAS is defined as individuals who have received at least one injection of the study medication and have provided informed consent.
The Kaplan–Meier method will be employed to determine the survival curves for OS, PFS, and duration of response. The median and 95% confidence intervals (CIs) are calculated at the same time. ORR and DCR were analyzed by descriptive statistics, and 95% CIs were calculated by the Clopper–Pearson method. Univariate and multivariate Cox analyses were performed to analyze the effects of patients’ baseline characteristics on PFS and OS. The safety analysis is based on the SAS and encompasses safety indicators such as the incidence of all AEs, the incidence of TRAEs, and the incidence of SAEs.
Discussion
In second-line treatment of cholangiocarcinoma, even with the opportunity to choose targeted therapies, patients are deterred by their high prices. For the vast majority of patients without specific targetable mutations, chemotherapy remains the primary treatment option. As immunotherapy advances, adjustments to second-line regimens are increasingly influenced by the outcomes of prior clinical studies. In 2022, the results of TOPAZ-1 trial 14 indicated that the combination of durvalumab which is a PD-L1 monoclonal antibody with GemCis significantly improved patient survival and was more effective than chemotherapy alone. Another trial, KEYNOTE-966 in 2023, also yielded positive results. 15 Consequently, chemotherapy is progressively being used in conjunction with immunotherapy as a second-line treatment for cholangiocarcinoma. This study is the first to evaluate the validity and harmfulness of cadonilimab combined with the NIFU regimen in locally advanced unresectable or metastatic cholangiocarcinoma patients.
Liposomal irinotecan, which utilizes liposomal technology, has a longer circulation time than conventional irinotecan, can specifically target tumor areas, and it offers better efficacy with fewer adverse reactions. 16 A randomized, controlled, phase IIb clinical trial in advanced biliary tract cancer, the NIFTY study7,8 revealed that the second-line NIFU regimen substantially improved the efficacy of advanced biliary tract cancer treatment. The ORR reached 12.5%, which was substantially superior to the control group that of fluorouracil plus calcium folinate (3.5%, p = 0.04), and the survival time significantly increased. The regimen containing liposomal irinotecan in the NIFTY study was superior in absolute terms to the FOLFOX regimen in the ABC-06 study. Although not a direct comparison, it still brings hope. Prior research has indicated that liposomal irinotecan has the potential to augment the anticancer properties of immune checkpoint inhibitors by elevating tumor neoantigen levels and enabling the identification of antigens by T cells. The combination of the two has shown better efficacy than using them separately.12,13
Cadonilimab is a type of dual-specific antibody that targets both PD-1 and CTLA-4. It was developed independently in China and has also been utilized in other clinical trials. Both CTLA-4 and PD-1 are negatively regulated signaling receptors of T-cell activation, but they act at different locations and times: CTLA-4 inhibits newly activated T cells in the early stages of immune circulation by binding to B7 on dendritic cells (antigen-presenting cells) in lymph nodes. 15 By contrast, PD-1 inhibits effector T cells in peripheral tissues or tumor sites, regulating immune responses. 17 Therefore, the combination of CTLA-4 and PD-1/PD-L1 could enhance the efficacy of immunotherapy. 18 Previous research has demonstrated that dual immunotherapy regimens, which incorporate both PD-1 or PD-L1 inhibitors and CTLA-4 inhibitors, have superior efficacy compared to the use of PD-1 or PD-L1 inhibitors alone. 12 Cadonilimab has been approved as a second-line monotherapy for the second-line treatment of advanced cervical cancer which is recommended by the National Comprehensive Cancer Network. A recent phase II clinical trial presented at the 2023 European Society for Medical Oncology Congress demonstrated that the combination of 6 mg/kg cadonilimab and lenvatinib as a first-line treatment for advanced liver cancer resulted in a significant objective response rate (ORR) of 35.5%. The treatment also showed a median PFS of 8.6 months and a lengthy median OS of 27.1 months, with handleable side effects. 19
It is exploratory for this trial to combine the NIFU regimen with cadonilimab for patients with advanced cholangiocarcinoma to prolong survival. Moreover, the present trial aims to identify potential biomarkers related to treatment efficacy to provide powerful evidence for clinical decision-making.