
Abstract
Parathyroid carcinoma (PC) is extremely rare in children and adolescent. PC is more often sporadic, but also it could be associated with germline mutations. The clinical features of primary hyperparathyroidism (PHPT) are nonspecific in children and adolescent, which delays the diagnosis for years. This case of PC in a pediatric patient, caused by germline heterozygous pathogenic variant in exon 1 of the CDC73 gene (c.70 G > T, p. Glu24Ter) is the first to be reported in Russia. Due to the rarity of pediatric parathyroid malignancy, the diagnosis of this endocrine neoplasm remains a challenge. The main difficulties that we faced in the management of the patient were the morphological confirmation of diagnosis, multiple surgical interventions, and disseminated PC metastases. We describe a 13-year-old girl with delayed diagnosis of PC and subsequent local recurrence after several surgeries, who underwent specific radiation therapy that allowed controlling hypercalcemia.
Background
Primary hyperparathyroidism (PHPT) is uncommon among children and adolescents with the frequency 2–5 cases per 100,000 and an equal sex ratio.1,2 The main causes in this age group include sporadic adenomas (up to 80%) and hyperplasia. 2 Genetic syndromes that are associated with multiple abnormal parathyroid glands (PTG) may account for 5–10% of all cases of PHPT, 3 and are especially suspicious in childhood. Parathyroid carcinoma (PC), as the underlying cause of PHPT in pediatric patients, is extremely rare. The exact etiology and pathogenesis of PC are unclear; it occurs more often sporadically. Several cases of PC have been reported in patients with a history of neck irradiation and end-stage renal disease. 4 There is a hypothesis that the carcinoma may originate from long-term adenoma or a hyperplastic PTG, though the potential for such malignant transformation remains questionable. Another form of PC is familial, which can occur in hyperparathyroidism-jaw tumor syndrome (HPT-JT) and familial isolated PHPT, single cases have been described in multiple endocrine neoplasia type 1 and 2A (MEN1 and MEN 2A). Pediatric patients with PHPT are more often symptomatic than adult, skeletal, and renal manifestations usually present at the time of diagnosis. 2 There are no specific clinical features, which allow distinct PC from benign adenoma. However, it can present as a palpable cervical mass compressing the surrounding structures. There is also evidence of nonspecific PC-associated symptoms in childhood and adolescence including neurological (irritability, depression, weakness, proximal myopathy); gastrointestinal (vomiting, nausea, diarrhea, pancreatitis); and cardiovascular (long QT interval) disorders. 5 We report a clinical case of a child with PC caused by germline heterozygous pathogenic variant in CDC73 gene who presented with severe symptoms of PHPT at the age of 6 years. This is the first case of pediatric PC described in the Russian population. The reporting of this case conforms to the CARE guidelines. 6
Case presentation
Up to the age of 6 years, the girl grew and developed normally. Furthermore, she started to complain of weakness, fatigue, and gait disturbance. During next 5 years, there were progressive muscle weakness, pain in the upper and lower extremities. X-shaped curvature of the legs has been noticed.
At the age of 12 years, the patient noted a sharp deterioration in her condition: severe muscle weakness and pain, worsening of hallux valgus. Besides, she lost 20 kg over 3 months. Ultrasound (US) of the thyroid gland revealed a nodular formation 29 × 17 × 17 mm at the upper pole of the left lobe. Six months later, she underwent the injury of the left hip. X-ray examination showed an inhomogeneous zone in the right ilium with an osteolytic area surrounded by a rim of sclerosis (31 × 23 × 24 mm), dual-energy X-ray absorptiometry (DEXA) was not provided. During a laboratory examination, PHPT was suspected (Table 1): an elevated parathyroid hormone (PTH) level 208 pmol/L (1.9–6.9) combined with hypophosphatemia 0.9 mmol/L (1.45–1.75), highly normal total calcium (Ca) 2.5 mmol/L (2.15–2.55), deficient of 25(OH)vitamin D – 8.0 ng/mL (>30), and extremely high alkaline phosphatase (ALP) 2105 U/L (141–460).
Laboratory test results and treatment.

Cinacalcet 30 mg/day, cholecalciferol 7000 IU/day
ALP, alkaline phosphatase; HTE, hemithyroidectomy; IHC, immunohistochemical; PC, parathyroid carcinoma; PTE, parathyroidectomy; PTG, parathyroid gland, TTF-1, thyroid transcription factor 1.

Parathyroid cancer (primary tumor). (a) PC’s cells with slightly eosinophilic cytoplasm and well-developed vasculature; the perinuclear rims of enlightenment are clearly visible. (b) Mitosis (→); large nucleus, a preserved nuclear–cytoplasmic ratio and perinuclear rims of enlightenment. (c) Intense expression of PTH. (d) Loss of parafibromin expression.

Recurrent PC. (a) Invasive growth in soft tissues and skeletal muscles. (b) Loss of parafibromin expression.
The patient underwent selective parathyroidectomy (PTE) of the altered left upper PTG at the age of 13 years. Initially, during morphological examination, the tumor was regarded as PTG adenoma (Table 1). Laboratory data of the early postoperative period are not available.
A few months later, lab tests showed elevated PTH levels [up to 122.2 pg/mL (15–65)], as well as normocalcemia [Ca total 2.34 mmol/L (2.1–2.55)], hyperphosphatemia [1.86 mmol/L (1.45–1.78)] and reduced urinary Ca excretion with normal serum creatinine level (Table 1). US scan performed by an expert-level specialist did not detect pathologically altered PTG in the typical places. Thus, secondary hyperparathyroidism was suspected. Calcium carbonate 1000 mg/day and alfacalcidol 0.5 μg/day were prescribed following the decrease of the PTH level to 107.2 pg/mL (15–65) with normocalcemia maintained.
To specify the cause of PHPT in pediatric patient, the next-generation sequencing using the panel of nine genes (MEN1, CASR, CDC73, CDKN1A, CDKN1B, CDKN1C, CDKN2A, CDKN2C, CDKN2D) was carried out. We performed sequencing at Illumina MiSeq sequencer (Illumina, USA) and applied paired-end reads (minimum coverage depth × 100) strategy. Genomic coordinates refer to the latest version GRCh38. The genetic study demonstrated germline heterozygous pathogenic variant c.70 G > T, p. Glu24Ter in exon 1 of the CDC73 gene (Figure 3) which was previously described in PC. 7 There is no family history of bone deformities, low-energy fractures, jaw tumors, or kidney disease (kidney cysts).
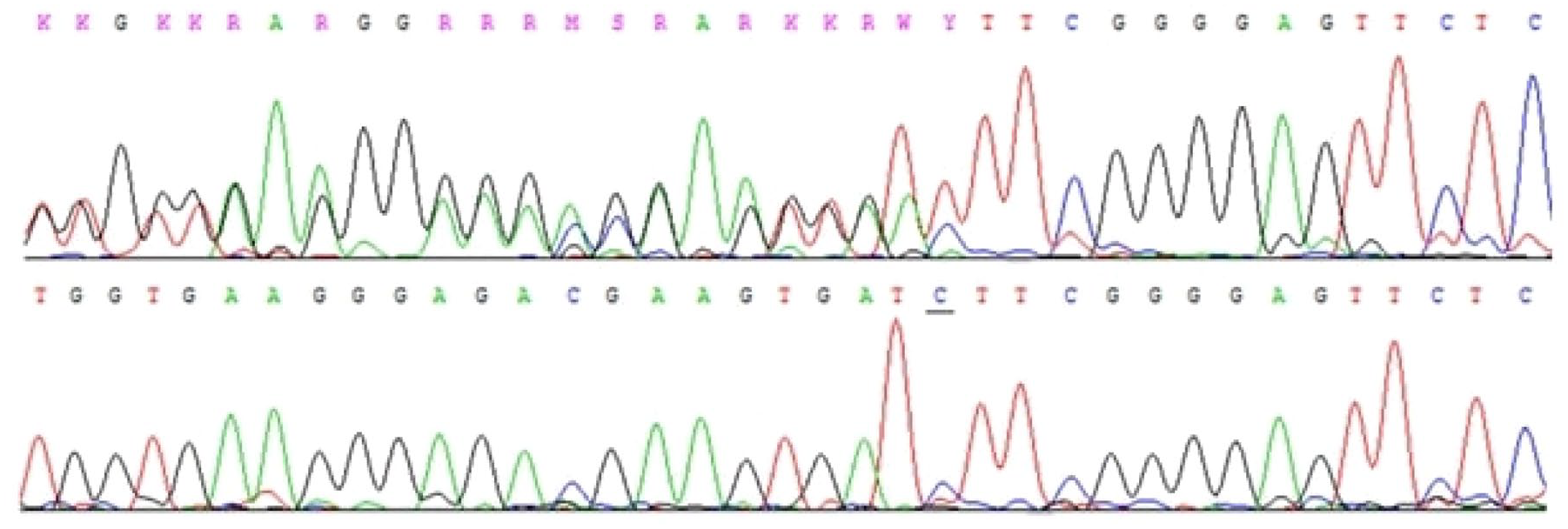
Chromatogram of exon 1 of the CDC73 gene. Germline heterozygous pathogenic variant c.70 G > T, p. Glu24Ter in exon 1 of the CDC73 gene (top) and normal sequence (bottom).
Relapse of PHPT happened at the age of 14 years: PTH 762 pg/mL (15–65), Ca ionized 1.4 mmol/L (1.03–1.29), total Ca 3.05 mmol/L (2.1–2.55) (Table 1). The leading complaints were severe osteoarticular and muscle pain (Table 1). X-ray revealed no fractures, valgus deviation of the knee joints with upward and outward rotation of the patella, Looser zones, and cysts of 2–9 mm in the medial epicondyles of the femurs. We detected nephrocalcinosis in the right kidney. US scan, as well as 99mTc-MIBI scintigraphy with single-photon emission computed tomography combined with computed tomography (SPECT-CT) revealed a lesion 8 × 4 × 6 mm at the posterior surface of the left thyroid lobe. Given the suspicion of a malignant tumor, surgeons removed a fragment of paratracheal tissue and thymus horn with areas of scars and muscles in addition to the altered PTG (Table 1). Histopathologic and immunohistochemical (IHC) examination verified the PC (Table 1, Figure 2). Moreover, the revision of histological slides after the first operation confirmed the vascular invasion (primarily regarded as a consequence of histologic processing) (Table 1). The diagnosis of PC was finally confirmed. In the early postoperative period, the patient developed hypocalcemia [total Ca 1.96 mmol/L (2.1–2.55), Ca ionized 0.99 mmol/L (1.03–1.29)] requiring Ca and alfacalcidol supplementation. However, the PTH level varied from 68.8 to 124 pg/mL (15–65).
Two years later, the disease recurred (Table 1). US scan revealed altered cervical lymph nodes – heterogeneous structures with increased blood flow on both sides 3 × 2, 6 × 3, 9 × 5, 13 × 6, and 12 × 4 mm. Metastases were confirmed by parathyroid fine needle aspiration (FNA) with PTH washout of the left and right lymph nodes (1732 and 3273 pg/mL, respectively). The extended surgery [left hemithyroidectomy (HTE) with ipsilateral lymphadenectomy, partial excision of the muscles, tissue, and fascia of the anterior surface of the neck] did not lead to the biochemical remission: PTH 149 pg/mL (15–65), total Ca 2.7 mmol/L (2.15–2.55). The 18F-FDG and 18F-choline positron emission tomography/CT imaging did not reveal any zones of increased accumulation. However, US scan and 99mTc-MIBI scintigraphy with SPECT-CT showed multiple foci in the right anterior cervical and paratracheal lymph nodes. At follow-up the level of PTH and total Ca increased up to 253 pg/mL (16–65) and 2.8–3.05 mmol/L (2.15–2.55), respectively. We initiated therapy with Cinacalcet at a dose of 30 mg/day with titration up to 60 mg.
Taking into account the progression of the disease at the age of 17 years, 2 more operations were undertaken: a thyroidectomy with the removal of pretracheal lymph nodes and tissue on the right side followed by right lymphadenectomy of supraclavicular neck nodes (Table 1). However, there was no disease remission. Multiple surgeries have not resulted in complications (hoarseness of voice, severe neck scarring).
In October 2020 (17 years old), we were forced to increase the dose of Cinacalcet to 90 mg/day: PTH – 244.3 pg/mL (15–65), total Ca – 2.69 mmol/L (2.15–2.55), Ca ionized – 1.29 mmol/L (1.03–1.29), phosphorus (P) – 1.98 mmol/L, 24-h urine Ca – 12.6 mmol/day (2.5–8) (Table 1). US scan showed an intermuscular hypoechoic lymph node 9 × 12 × 8 mm most likely corresponding to a metastasis in the projection of the removed right PTG (in the lower third of the neck). This was inconsistent with the results of 99mTc-MIBI scintigraphy with SPECT-CT and multislice computer tomography (MSCT) that demonstrated a low isotope uptake of an altered lymph node 12 × 10 × 10 mm in the subcutaneous fat on the front neck on the right. No hyperactive foci were identified on the SPECT in the ‘whole body’ and ‘pelvis’ modes.
Given the ineffectiveness of the previous five surgical interventions, the patient was referred to radiotherapy on the neck area followed by local irradiation of the metastatic lesion on the neck (identified on MSCT in the specialized oncological institution) (Table 1). During 3 years of follow up after radiotherapy, the level of PTH slowly got down that allowed to decrease dose of Cinacalcet from 90 to 60 mg/day (Table 1). Throughout the follow-up, bone mineral density of the spine, proximal femur, and radius, according to X-ray densitometry, remained within normal values.
US scan in February 2021 showed a hypoechoic avascular formation of irregular shape 15 × 8 × 10 mm on the anterior surface above the postoperative scar and another one near the jugular notch 12 × 6 × 10 mm. At the same time, these lesions did not accumulate 99mTc-MIBI on SPECT-CT. US scans visualized a hypoechoic formation with uneven contours in the projection of the thyroid cartilage, dimensions 5 × 2.5 × 3 mm without blood flow (probably a cyst), normal lymph nodes. However, there were no volumetric formations on CT with contrast enhancement.
In March 2022 (18 years old), emergency mini-percutaneous nephrolithotripsy with nephrostomy on the right was performed. Postoperative MSCT of the kidneys showed small stones in the pelvis of the right kidney up to 4 mm, in the upper group of calyces of the left kidney up to 3 mm.
In October 2023 (20 years old), after discontinuation of Cinacalcet for 7 days: albumin adjusted Ca 2.63 mmol/L (2.15–2.55), PTH 104 pg/mL (15–65). Therapy with Cinacalcet 30 mg/day was continued. US scan of the neck and MSCT with contrast revealed reduction of the largest lesion to 6.5 × 7 × 5.5 mm (June 2020 – 12 × 10 × 10 mm). Screening for complications of PHPT was carried out: estimated Glomerular filtration rate using Chronic Kidney Disease Epidemiology Collaboration equation (eGFR (CKD-EPI)) 131 mL/min/1.73 m 2 ; no signs of nephrolithiasis/nephrocalcinosis were detected on US. DEXA demonstrated increase in bone density to the Z-score in L1–L4 −1.1 SD (+11.9% compared to October 2020), left femoral neck 0 SD (+14.9% compared to October 2020), radius −1.7 SD (+43.6 compared from July 2017).
Genetic study did not reveal pathogenic variants of CDC73 gene in parents. The patient has an 11-year-old younger brother who does not undergo genetic study, but regularly tests for Ca and PTH remain normal.
Discussion
About 20 cases of PC under the age of 16 years have been previously published (Table 2).7–26 PC is more often sporadic. In this case, the first mutation occurs de novo (first stroke) followed by a stepwise inactivation of the second allele in the same PTG somatic cell (second stroke) which leads to clonal tumor growth. Thus, the inheritance of a germline mutation at birth explains the earlier age of PHPT manifestation, which is more typical for familial forms, while sporadic ones has a later onset of the disease. Such mechanism is consistent with Knudson’s two-hit hypothesis.
PC in children from 1972 to 2022.

HTE, hemithyroidectomy; mts, metastases; ND, no data; PC, parathyroid carcinoma; PTE, parathyroidectomy; PTH, parathyroid hormone.
Mutations in the CDC73 gene play a crucial role in the pathogenesis of PC. The CDC73 gene consists of 17 exons located on chromosome 1q31.2 and encodes the parafibromin protein associated with the Paf1 protein complex (polymerase I-associated factor). 27 HPT-JT (OMIM#145001) is a rare syndrome characterized by PTG tumors, fibro-osseous lesions (ossifying fibroma) of the mandible or maxilla, cystic and neoplastic renal abnormalities. PHPT (in 15–20% due to carcinomas) is usually the first manifestation of the disease and occurs in >90% of HPT-JT cases. Germline pathogenic variants in the CDC73 gene are present in about 90% of patients with HPT-JT and in one-third of patients with sporadic PC with no family history. 28 The earliest case of hypercalcemia in a patient with HPT-JT was recorded at the age of 7 years, 29 and the latest in the sixth decade of life. 30 The median age of PHPT debut is 27 years (range 12–58).31–33 In the presented case, clinical manifestation of PHPT was noted at the age of 6 years, but the biochemical evaluation was performed only 6 years later. We did not confirm other components of the HPT-JT syndrome.
Due to the lack of reliable preoperative diagnostic criteria, the diagnosis of PC is based on histological and IHC examinations. According to the 2022 World Health Organization (WHO) classification of endocrine and neuroendocrine tumors, the histological definition of PC still requires one of the following findings: angioinvasion (vascular invasion), lymphatic invasion, perineural (intraneural) invasion, local malignant invasion into adjacent anatomic structures, or histologically/cytologically documented metastases. 34 In the absence of invasive growth, but in the presence of suspicious signs for malignancy (broad fibrous bands, mitoses, adherence to surrounding structures, monotonous sheet-like or trabecular growth pattern, nuclear atypia) atypical parathyroid tumor is diagnosed. 34 An additional diagnostic method is an IHC analysis including oncomarkers expression. PC is associated with negative expression of parafibromin, Adenomatous Polyposis Coli (APC), E-cadherin, p27, Bcl-2a, mdm-2, and 5-hmC, and positive for PGP9.5, galectin3, human telomerase reverse transcriptase (hTERT), as well as p53 overexpression and increased Ki-67 (often >5%). The term ‘parafibromin deficient parathyroid neoplasm’ has been introduced in the 2022 WHO classification for the first time. The loss of parafibromin expression in the presence of internal positive controls (fibroblasts, endothelial cells) is an indication for genetic testing for CDC73 pathogenic variant. 34 Unfortunately, genetic study and IHC have limited availability in routine practice.
The difficulties of morphological diagnosis are clearly presented in the presented case. The primary tumor was not initially regarded as a ‘carcinoma’. However, in 2016, the fifth edition of the WHO classification of tumors of the endocrine system drew the attention of pathologists to the difficulties of morphological diagnosing of malignant tumors and put forward such a subgroup of tumors as atypical. Morphological diagnosis after second surgery also caused difficulties. Taking into account that the primary tumor was regarded as an ‘adenoma’ and the focus of recurrence was localized at the site of the primary operation, it was necessary to exclude the possibility of continued growth of residual PTG tissue. However, the invasion in skeletal muscles through ‘tongues of flame’ was suspicious for carcinoma recurrence. Gill et al. 35 described similar morphological difficulties and a long way to a correct diagnosis in a 16-year-old boy with a mutation in the CDC73 gene and loss of parafibromin expression. In the presented case, the primary tumor showed a pronounced branched vasculature, large nuclei with a preserved nuclear–cytoplasmic ratio, and a perinuclear rim [Figure 1(a) and (b)]. Subsequently, the loss of parafibromin expression in the primary tumor (first operation) [Figure 1(d)] and in pattern of recurrence tissue (second operation) [Figure 2(b)] was confirmed. The results of the IHC study correlated with genetic testing, which revealed germline heterozygous pathogenic variant in the CDC73 gene.
The main treatment option for both primary and secondary foci of PC is surgery. There are still debates on the preferred volume of primary surgery in patients with a CDC73 mutation: bilateral revision of the neck with subtotal/total PTE or selective operation. 36 Previously, preventive subtotal/total PTE remains the optimal strategy to reduce the risk of PC recurrence. However, this approach is associated with the high risk of postoperative hypoparathyroidism. Some authors suggested autotransplantation of PTG to avoid it.37,38 However, autotransplantation of PTG tissue in patients with CDC73 pathogenic variant may lead to dissemination of malignant cells. 39
Routine cytological study of PTG formations is not recommended, since it does not allow differentiating benign from malignant lesions and can promote the spread of malignant cells along the tract of the puncture needle. 40 In our case, FNA could also contribute to tumor progression.
Five-year survival rate is 76–85%, and 10-year survival varies from 49 to 77%. 41 Overall survival depends on multiple factors, including the type of primary surgery (i.e. PTE or en bloc resection), serum Ca levels and/or presence of resistant hypercalcemia, local or distant metastases, CDC73 mutation, and/or loss of parafibromin or Ca-sensing receptor protein expression. Besides hormonally inactive PC has poor prognosis. 42 Complications associated with hypercalcemia are the main cause of death. 43 Relapse of the PC develops in 50% of cases, a third of them are regional metastases. Distant metastases are detected in 25% of cases, more frequently in the lungs (40%) and liver (10%), less often in the bones, pleura, pericardium, and pancreas. 44 For already developed metastases, the 5-year survival rate reduced to 50%.45–47 Surgery remains the main treatment for PC recurrence; however, decision on its volume is made individually and depends on technical possibilities.48–50 ‘Aggressive’ surgical approaches for recurrent disease were associated with 30% increase in overall survival.51,52 Other treatment options for PC metastases are still challenging since there are no effective regimes of chemo- or radiotherapy. 53 There is a relatively successful experience of radiotherapy as an adjuvant treatment after surgery in locally invasive PC, in cases of microscopic residual disease or after multiple recurrences, but the conclusions of the study were ambiguous and based on a small sample of patients.53–55 The American Association of Endocrine Surgeons guidelines 56 recommends reserving external radiotherapy for palliative treatment because surgery on a radiated field may be difficult. Therefore, radiotherapy may be considered for refractory disease in patients who are not candidates for re-operation.
Several cases of successful anti-PTH immunotherapy have been presented, which led to long-term control of hypercalcemia and tumor growth, and in some cases to decrease in the size of metastases.57–59 Some authors described a successful experience of the multikinase inhibitor therapy with sorafenib for metastatic PC foci in the lungs. 60 However, the targeted therapy can be only applied if the tumor progresses and the size of the secondary lesions is appropriate.
In our case, due to the ineffectiveness of the multiple surgical interventions, the remote radiation therapy followed by local irradiation of the metastatic lesion was performed to attempt stop progression. Three years of follow-up showed us promising results of slowing decrease of level of PTH and Ca (Table 1). Further observation will allow drawing conclusions. However, the remote radiation therapy is a possible way to stabilize the aggressive course of PC, if surgical treatment is ineffective or impossible.
Conclusion
PHPT is not common in the pediatric population but often has aggressive clinical behavior. This indicates the mandatory screening for Ca and PTH levels in children with bone or kidney disorders for earlier verification of the disease. Moreover, early onset of PHPT requires the exclusion of hereditary forms. If there is a pathogenic variant in the CDC73 gene, doctors should be wary of PC and possibly an ‘aggressive’ surgical approach. Alternative treatment options such targeted and radiation therapy may be considered if surgery is ineffective.
Supplemental Material
sj-pdf-1-tam-10.1177_17588359241265222 – Supplemental material for Combination approach for CDC73-related parathyroid carcinoma in an adolescent female patient: a case report and literature review
Supplemental material, sj-pdf-1-tam-10.1177_17588359241265222 for Combination approach for CDC73-related parathyroid carcinoma in an adolescent female patient: a case report and literature review by Ekaterina Kim, Natalia Kalinchenko, Anna Eremkina, Liliya Urusova, Rustam Salimkhanov and Natalia Mokrysheva in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.