
Abstract
Pancreatic cancer is one of the deadliest malignancies in humans and it is expected to play a bigger part in cancer burden in the years to come. Pancreatic ductal adenocarcinoma (PDAC) represents 85% of all primary pancreatic malignancies. Recently, much attention has been given to PDAC, with significant advances in the understanding of the mechanisms underpinning disease initiation and progression, along with noticeable improvements in overall survival in both localized and metastatic settings. However, given their rarity, rare histological subtypes of pancreatic cancer have been underappreciated and are frequently treated as PDAC, even though they might present non-overlapping molecular alterations and clinical behavior. While some of these rare histological subtypes are true variants of PDAC that should be treated likewise, others represent separate clinicopathological entities, warranting a different therapeutic approach. In this review, we highlight clinical, pathological, and molecular aspects of rare histological types of pancreatic cancer, along with the currently available data to guide treatment decisions.
Plain language summary
The most common type of pancreatic cancer is ductal adenocarcinoma. While much attention has been given to the molecular aspects and treatment aspects of this disease, rare variants of pancreatic cancer have been underappreciated. Some of them present unique molecular features that suggest different treatment approaches could lead to better outcomes. In this review, we summarize information on the clinical, pathological, and molecular features of rare subtypes of pancreatic cancer, along with subtype-specific data on treatment.
Introduction
Pancreatic cancer is currently the 12th most incident malignant neoplasm and the 6th most common cause of cancer-related death worldwide. 1 In the USA, pancreatic cancer ranks 10th and 7th among the most frequent cancers among men and women, respectively. 2 Additionally, in women pancreatic cancer has surpassed colorectal cancer as the third most common cause of cancer-related death among women. Indeed, data from developed and developing countries have systematically predicted an increasing epidemiological burden from pancreatic cancer in the years to come.3–5
Pancreatic ductal adenocarcinoma (PDAC) represents 85% of all malignant pancreatic tumors. 6 For this tumor, current guidelines [e.g. National Comprehensive Cancer Network and European Society of Medical Oncology (ESMO)] support the use of polychemotherapy regimens such as FOLFIRINOX and gemcitabine plus nab-paclitaxel in multiple clinical scenarios based on the results of randomized clinical trials (RCTs) that showed improved survival when compared to single-agent gemcitabine.7,8 While PDAC has been given proper attention given its relative chemoresistance and increasing epidemiological burden, rare variants of pancreatic cancer have been underappreciated as patients with such tumors have been systematically excluded from clinical trials of pancreatic cancer. 9 However, recent data suggest the incidence of some of these variants has increased at a faster pace than that of PDAC. Additionally, molecular analyses have shown some of these tumors have specific molecular alterations that segregate them from PDAC, suggesting that at least some of the lessons learned from studies of PDAC might not be automatically transferred to the treatment of patients with rare histological variants of pancreatic cancer.
In this narrative review, we explore clinical, pathological, and molecular data specific to these rare variants of pancreatic cancer to help understand histology-tailored treatment. On 1 December 2023, we searched PubMed from 2013 onward using descriptors for rare variants of pancreatic cancer and employed backward reference listing to conduct and write this narrative review. We also looked for relevant abstracts presented at the American Society of Clinical Oncology and ESMO annual meetings. We made no restrictions based on language, publication status, or type of manuscript. We didactically distinguish variants of PDAC according to the World Health Organization (WHO) Digestive System Tumor Classification from other epithelial malignancies arising in the pancreas. Table 1 outlines key clinical and pathological aspects of these rare pancreatic cancer subtypes. Table 2 summarizes the role of different treatment modalities in the management of these tumors, rating the strength of recommendation according to the GRADE handbook. 10
Key clinical, pathological, and molecular data of rare histological subtypes of pancreatic cancer.
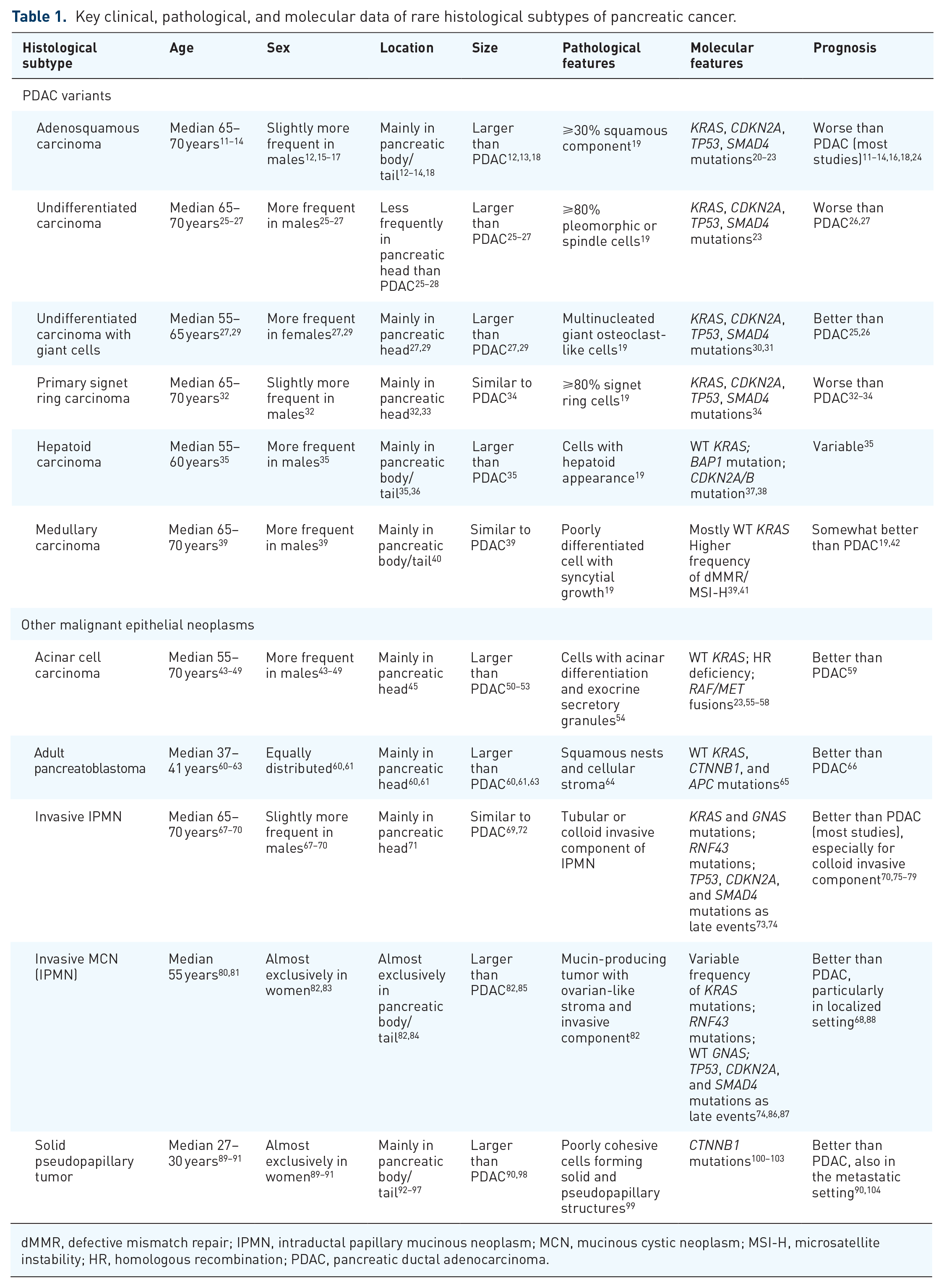
dMMR, defective mismatch repair; IPMN, intraductal papillary mucinous neoplasm; MCN, mucinous cystic neoplasm; MSI-H, microsatellite instability; HR, homologous recombination; PDAC, pancreatic ductal adenocarcinoma.
Summary of the role of different treatment modalities in the management of rare histological subtypes of pancreatic cancer.

Strength of recommendation is graded according to the GRADE handbook. 10
AP, adult pancreatoblastoma; dMMR, defective mismatch repair; FFX, FOLFIRINOX; GEM + NAB, gemcitabine plus nab-paclitaxel; HRR, homologous recombination repair; IPMN, intraductal papillary mucinous neoplasm; MSI-H, microsatellite instability; PDAC, pancreatic ductal adenocarcinoma; TMB, tumor mutational burden.
One should highlight that the vast majority of data on treatment of these rare variants of pancreatic cancer are derived from retrospective studies, in many of which treatment end-point was not the primary focus. Also, in some of these investigations, patients have been treated with treatment schedules not considered to be standard-of-care by today’s standards given that many patients were treated before the results of RCTs using the modern polychemotherapy regimens (FOLFIRINOX and gemcitabine plus nab-paclitaxel) were available.
Variants of PDAC
Pancreatic adenosquamous carcinoma
Pancreatic adenosquamous carcinoma (PADSC) is defined by the WHO Digestive System Tumor Classification as a histological subtype of ductal adenocarcinoma in which the squamous cell component arbitrarily corresponds to ⩾30% of the neoplasm. 19 It represents 1–4% of all malignant pancreatic neoplasms 180 and recent data suggest an increasing incidence of this histological variant over the past two decades. 181 The frequency of this subtype is greater in surgical series given the difficulty in identifying the malignant squamous cells in samples of fine-needle aspiration (FNA). 182
As squamous cells are not found in healthy pancreatic tissue, many theories have tried to explain the existence of PADSC. One model suggests pancreatic ductal epithelial cells undergo squamous metaplasia as a result of chronic inflammatory stimuli (squamous metaplasia theory). 183 Alternatively, the same progenitor cell might be responsible for the adenocarcinoma and squamous carcinoma components (differentiation theory). This concept is supported by the fact that the squamous cell carcinoma and the adenocarcinoma components harbor similar genomic alterations. 21 Indeed, recent phylogenetic studies indicate that the squamous cell features represent a subclonal phenomenon associated with additional genetic and epigenetic events. 184
Patients with PADSC present similar age and sex distribution when compared to PDAC.11–14 Moreover, levels of the tumor markers CA 19-9 and carcinoembryonic antigen (CEA) are alike between the two histologies. 18 At presentation, PADSCs are more likely than PDACs to be large12,13,18 or high-grade tumors13,14,18 and more frequently are locally in the pancreatic body/tail.12–14,18 Some data also suggest PADSCs are more frequently associated with macro 18 or microvascular invasion 14 and regional lymph node metastases. 18 The clinical presentation of PADSC is similar to that of PDAC, with most patients presenting abdominal pain, nausea/vomiting, bloating, weight loss, and jaundice. 106
On cross-sectional imaging, PADSCs and PDACs have similar radiological features. Foci of squamous cell carcinoma are more likely to undergo necrosis and studies of computed tomography (CT) and magnetic resonance imaging (MRI) suggest focal necrosis is more frequent in PADSC than in PDAC.185,186 In this sense, due to its improved soft tissue contrast, MRI can detect focal necrosis more accurately than CT. 186 Additionally, PADSCs are more likely to present peripheral ring enhancement.185,186
PADSC typically presents as a sizable and firm mass, characterized by ill-defined and infiltrative edges. 19 Squamous differentiation within pancreatic carcinomas is relatively rare and typically occurs alongside conventional ductal adenocarcinoma. Squamous differentiation manifests as solid clusters or layers of polygonal cells with well-defined cell borders, prominent intercellular junctions, dense eosinophilic cytoplasm, and varying degrees of keratinization. 19 In cases of pure squamous cell carcinoma, metastasis from other sites, especially the lungs, should be ruled out.
Immunohistochemically, PADSCs display loss of expression of the p16 protein (CDKN2A), loss of the SMAD4 protein (DPC4), and strong nuclear p53 immunoreactivity, akin to the molecular signature found in PDACs. 187 Also, the squamous component often expresses p63, p40, and low molecular weight cytokeratins. The squamous component of PADSC has a higher proliferative rate, as shown by the increased ki-67 staining on immunohistochemistry. 188 Additionally, a subset of PADSC expresses PD-L1, (programmed death ligand 1), which is restricted to the squamous cell carcinoma component.189,190
In resemblance to PDAC, PADSCs commonly have mutations in KRAS (73–100%), CDKN2A (6–52%), and SMAD4 (18–26%).20–23 However, TP53 mutations might be more common in PADSC (85–88%) than in PDAC (60–70%). 191 Additionally, a high frequency of chromosome 3p loss has been described in some cohorts of patients with PADSC. 21 PADSCs’ areas with squamous features or differentiation display high rates of mutations in chromatin modifier genes and MYC amplification when compared to PDAC, suggesting that epigenetic dysregulation secondary to chromatin modifier gene mutations and downstream effects of MYC activation might contribute to the squamous cell phenotype.22,184 Also, almost all cases of PDAC with squamous features or differentiation and PADSC present gene expression patterns compatible with Bailey’s squamous or Collisson’s quasi-mesenchymal subtypes. 184
In the localized setting, surgical resection has been associated with improved survival for patients with PADSC.11,14,18 Additionally, most series suggest adjuvant chemotherapy leads to improved survival.14,18,105–108 Interestingly, limited data indicate platinum-based chemotherapy (either FOLFIRINOX or cisplatin/oxaliplatin plus gemcitabine/capecitabine) might be more effective against PADSC in the adjuvant setting. 108 Also, population-based studies and a retrospective single-institution analysis suggest patients with resected PADSC submitted to adjuvant chemoradiotherapy have better overall survival (OS),105,107,109 possibly secondary to the better locoregional control associated with radiotherapy for larger and more locally aggressive tumors such as PADSCs. Finally, either neoadjuvant therapy or upfront surgery can be considered acceptable options for patients with resectable PADSC. 107 However, as for PDAC, patients with borderline resectable or locally advanced PADSC are best treated with neoadjuvant chemotherapy followed by surgery.192,193
Chemotherapy has also been associated with improved survival in the metastatic setting. 110 Limited data suggest combination chemotherapy leads to better overall response rate (ORR), disease control rate (DCR), progression-free survival (PFS), and OS. 17 Newer chemotherapy regimens, such as FOLFIRINOX and gemcitabine plus nab-paclitaxel are more active than older ones. 15 In one multi-institutional series from Asia, FOLFIRINOX and gemcitabine plus nab-paclitaxel displayed similar results in terms of ORR (20.0% versus 26.9%; p = 1.000), DCR (60.0% versus 46.1%; p = 0.468), PFS (2.3 versus 2.8 months; p = 0.476), and OS (7.2 versus 7.3 months; p = 0.887). 17 Equivalence in OS between FOLFIRINOX and gemcitabine plus nab-paclitaxel was also seen in a multicentric French study (11.8 versus 15.4 months; p = 0.500). 15 Therefore, either FOLFIRINOX or gemcitabine plus nab-paclitaxel seems to be reasonable in the first-line treatment of patients with metastatic PADSC.
PADSCs are almost always microsatellite stable and have a low tumor mutational burden (TMB), with a mean of 4.7 Mut/Mb. 193 This suggests the use of checkpoint inhibitors against these tumors might not be effective. However, given that a subset of PADSC displays PD-L1 expression, the use of anti-PD-1/PD-L1 monoclonal antibodies seems rational. In one case report with high PD-L1 expression, the use of sintilimab, an anti-PD-1 agent, in combination with gemcitabine, led to a sustained partial radiological response. 194
Interestingly, a handful of clinical trials specifically designed for patients with PADSC are ongoing. In phase II clinical trial, minnelide, an oral anti-super-enhancer drug that inhibits MYC expression in preclinical models of PADSC is being tested (NCT03129139). In another phase II trial, patients with microsatellite-stable PADSC are to receive the anti-PD-1 agent retifanlimab as a single-agent (NCT04116073).
Some reports suggest patients with PADSC have worse clinical outcomes than those with PDAC, both in the localized and metastatic setting.12–14,16 However, there is some controversy on this matter, as other studies have failed to describe inferior survival for patients with PADSC.11,18,24 Among patients with localized PADSC, location in the head of the pancreas, high levels of CA 19-9 or CA 125, blood vessel invasion, multifocal disease, and lack of adjuvant chemotherapy predict a worse prognosis.14,18,107,195 In the metastatic setting, (Eastern Cooperative Oncology Group (ECOG) performance status ⩾2, synchronous metastases, lung or peritoneal metastases, and elevated levels of CA 19-9 or C-reactive protein (CRP) are associated with worse survival.15,17 Interestingly, the amount of squamous cell carcinoma in PADSC has not been associated with inferior survival. 105
Undifferentiated carcinoma of the pancreas
Undifferentiated carcinomas of the pancreas (UCPs) are tumors in which a substantial part does not show a definitive direction of differentiation. 19 Collectively, this heterogenous group of tumors is difficult to define in terms of epidemiological and clinical characteristics, as well as predictive and prognostic information, due to the dearth of clinical data and the recent changes in their pathological classification. It has been estimated that UCPs represent 1–7% of all pancreatic cancers. 180 Under the current WHO Digestive System Tumor Classification, UCPs are classified into three morphological subtypes: anaplastic undifferentiated carcinoma of the pancreas (AUCP), sarcomatoid undifferentiated carcinoma of the pancreas (SUCP), and carcinosarcoma of the pancreas (CSP). 19 Additionally, a subset of undifferentiated carcinomas presents osteoclast-like multinucleated giant cells admixed with histiocytes and neoplastic cells and have been described as undifferentiated carcinomas with osteoclast-like giant cells of the pancreas (UCOGCP). The latter subtype is associated with improved clinical outcomes and, therefore, is considered a distinct clinical entity.
While most UCOGCPs arise in the pancreatic head, other types of UCP are less likely to occur in the pancreatic head than PDAC.25,27,29 Also, these tumors are larger than PDACs at the time of diagnosis, usually measuring more than 5 cm. Except for UCOGCPs, UCPs are more common in males.27,29 The clinical presentation is similar among different subtypes of UCP. 27 Patients frequently present abdominal pain, weight loss, and hyporexia.27,29,112 In contrast to PDAC, jaundice is less frequently found in UPCs (<30% at presentation).27,29 CEA and CA 19-9 levels are elevated in 13.3–44.4% and 36.4–73.0% of cases, respectively.27,29
Hyperleukocytosis, severe anemia, and elevated serum CRP are frequently observed in patients with UPC. 196 It is suggested that these findings may be due to rapid tumor growth and intra-tumoral hemorrhage and necrosis and may also result from the production of granulocyte colony-stimulating factor.196,197 Besides the larger tumor size at presentation, UPCs are usually well-defined hypovascular masses associated with pancreatic duct dilatation.198,199 Necrosis and hemorrhage are also commonly described and foci of calcification due to heterologous sarcomatous component can be found.
AUCP (also known as pleomorphic carcinomas) are characterized by a combination of pleomorphic mononuclear cells admixed with abnormal (‘bizarre-appearing’) giant cells displaying eosinophilic cytoplasm. 19 The tumor comprises predominantly solid sheets of cells devoid of gland formation (at least 80%), showcasing highly variable nuclei. These tumors display a high frequency of epithelial–mesenchymal transition (EMT) activation, as shown by the lack of cell cohesion and the increased expression of EMT markers,200,201 such as E-cadherin loss induced by the E-cadherin promoter methylation. 202 The gene mutational profile of AUCP is similar to that of PDAC, with frequent mutations in KRAS (76%), TP53 (68%), and CDKN2A (40%). However, a somewhat lower frequency of SMAD4 mutations has been described. 23 Unlike PDAC, PD-L1 expression is frequently found in AUCPs (60%), 203 often with intra-tumoral PD-1-positive lymphocytes. Interestingly, this occurs despite the very low frequency of defective mismatch repair/microsatellite instability (dMMR/MSI-H) and the low median TMB (1.78 mutations/Mb). 23
SUCPs account for 1–2% of all pancreatic malignancies. 204 This tumor arises from the pancreatic epithelium and exhibits cells with a spindle cell morphology (⩾80%), potentially containing heterologous elements, such as bone and cartilage. 19 Although extremely rare, some particularly aggressive SUCPs have been noted to possess rhabdoid cells. 205 These tumors are identified by widespread sheets of rhabdoid cells, often demonstrating a lack of cohesion and embedded within a myxoid matrix. They may also present with pleomorphic giant cells, spindle cell areas, and tubular components. These tumors frequently present loss of members of the chromatin–remodeling switch/sucrose non-fermenting (SWI/SNF) complex,206,207 which is seen in immunohistochemistry as the absence of nuclear staining of SMARCB1 (INI1), a core subunit of the SWI/SNF chromatin–remodeling complex. In a recent analysis, SUCP has been found to harbor alterations in KRAS (mutation in 86–100%), TP53 (mutation in 86–90%), CDKN2A/B (loss in 18–40%), and SMAD4 (mutation in 10%) in similar frequencies to those found in PDAC.204,208 Additionally, KRAS amplification occurs in 7–30% of SUCPs. The TMB of SUCP is relatively low (median of 7.0 mutations/Mb), 204 with 2% of tumors being considered TMB-high (⩾10 mutations/Mb). 208 However, at least 60% of these tumors express PD-L1, suggesting immune checkpoint blockade might be a therapeutical option.
CSP are considered bona fide biphasic neoplasms, composed of mixed carcinomatous and sarcomatous components (with or without heterologous elements), normally separated without a transition zone. 209 Components displaying evident epithelial morphology can resemble conventional ductal adenocarcinoma. Arbitrarily, each component should constitute ⩾30% of the tumor to qualify the tumor as carcinosarcoma. 19 Importantly, each component typically demonstrates immunophenotypic similarities to its pure counterpart. 19 While the evidence regarding this entity is sparse, mainly consisting of case reports and small series, recent data suggest an increasing incidence of CSP, which now corresponds to 0.5% of pancreatic cancers. 210 Very limited data exist on the molecular characterization of CSP. Recently, the KRAS Q61H mutation was found in two CSPs. 211 Interestingly, analyses performed after laser capture microdissection have shown similar mutational (KRAS and TP53) and immunohistochemical (TP53, ARID1A, and KDM6A expression) profiles between carcinomatous and sarcomatous elements. However, higher Ki-67 expression was found in the sarcomatous component, which possibly reflects a more aggressive clinical behavior. 211
UCOGCP are rare pancreatic neoplasms consisting of three distinct cell populations: osteoclastic giant cells, histiocyte-like sarcomatoid carcinoma cells, and pleomorphic giant carcinoma cells.19,29 It corresponds to roughly 1% of all pancreatic malignant neoplasms. 29 Besides its unique morphological characteristics, UCOGCP can be clinically distinguished from other variants of UCP as it more commonly affects females and younger patients. 27 Macroscopically, cyst formation is a common feature, and hemorrhage or necrosis is typically observed within the tumor. 30 The multinucleated cells express histiocytic markers lack epithelial differentiation, and are often located near areas of hemorrhage or necrosis.212,213 The neoplastic cells display varied morphologies, ranging from spindle-shaped to epithelioid, with potential for being large and pleomorphic. They typically lack cohesion and may be found within the cytoplasm of the osteoclast-like giant cells. Also, UCOGCPs show a high propensity for intra-ductal growth and, surprisingly, a lesser frequency of regional lymph node metastasis when compared to PDAC. 29 Importantly, in contrast with other variants of UCP, the overall prognosis of UCOGCP seems to be better than that of PDAC (especially in the localized setting), with a subgroup of patients experiencing a rather indolent clinical course. 29
Supporting the current WHO classification as a PDAC variant, UCOGCP derives from ductal tumor clones and shares with PDAC the same genetic background, including a high frequency of activating mutations in the KRAS and inactivating mutations in CDKN2A, TP53, and SMAD4.30,31 Luchini et al. 30 described the presence of non-synonymous missense mutations in the same amino residue in SERPINA3, suggesting it as an oncogene. In the same study, mutations in GLI3 were found, suggesting that GLI3 is also a genetic driver of UCOGCP. dMMR/MSI-H is rarely found in UCOGCP (0–8%).214,215 However, similar to AUCP and SUCP, PD-L1 expression is frequent in UCOGCP (63–80%)203,214,215 and in this disease, PD-L1 expression has been linked to TP53 mutations 203 and inferior clinical outcomes.203,214
Surgical resection has been associated with improved survival in all subtypes of UCP,25–27,111 with the possible exception of anaplastic (pleomorphic) carcinoma.26,112 Also, currently there is no good quality data to guide the use of adjuvant chemotherapy for patients with UCP. Contrary to what one would expect, in some retrospective cohort studies long-term survivors have not received adjuvant chemotherapy.111,113 In a recent population-based study, Christopher et al. 26 found a non-statistically significant survival benefit for chemotherapy for those undergoing resection. Indeed, many of these patients were treated in an era when single-agent adjuvant chemotherapy was the standard-of-care for PDAC, and given the lower response rates of UCP when compared to PDAC, polychemotherapy should be considered standard in this setting. In the same analysis, adjuvant radiotherapy was associated with improved survival. 26 In a study evaluating exclusively patients with SUCP, Blair et al. 113 found both long-term survivors received radiotherapy. However, given the high propensity of UCP to recur at distant sites 113 and the lack of benefit of adjuvant radiotherapy for PDAC, we believe the routine use of radiotherapy in this setting is not warranted. Again, in cases of borderline resectable and locally advanced disease, neoadjuvant (or conversion) chemotherapy can lead to potentially curative resections.216–219
In the advanced setting, chemotherapy has been associated with improved survival. 26 So far, paclitaxel-based regimens seem to have the highest activity. In a multicenter retrospective cohort study of 50 patients with UPC treated with chemotherapy carried out in Japan, gemcitabine plus nab-paclitaxel provided a median PFS of 4.60 months and ORR of 33.3%. 114 The PFS times were 1.61 and 2.96 months and ORRs were 4.2% and 0% for gemcitabine and S-1, respectively. Gemcitabine plus nab-paclitaxel significantly improved PFS compared to gemcitabine (p = 0.014) and showed significantly higher ORR compared to gemcitabine or S-1 (p = 0.033 and p = 0.034, respectively). Furthermore, the first-line paclitaxel-containing regimen significantly improved OS (6.94 versus 3.75 months, p = 0.041) and was found to be an independent predictor of OS [HR: 0.221; 95% confidence interval (CI): 0.076–0.647; p = 0.006]. Similar data on paclitaxel-based therapy have been shown in small case series, 115 and a few complete responses to this regimen have been reported.116,117 As a result of these analyses, the Japanese group launched a clinical trial to prospectively investigate the activity of Gemcitabine plus nab-Paclitaxel for UCP (jRCTs031220099). On the other hand, the results of the activity of FOLFIRINOX against UCP are disputable. In a recent multi-institutional analysis from Japan, FOLFIRINOX and Gemcitabine plus nab-Paclitaxel showed similar activity. 23 Indeed, FOLFIRINOX has been associated with some clinical responses, 114 but limited data suggest a short-lived clinical benefit.116,220,221
Currently, there is no data on targeted therapy for UCP. However, the very high frequency of PD-L1 expression in UCP raises the possibility that immune checkpoint inhibitors (ICI) might be active. Indeed, anti-PD-1 monoclonal antibodies have been shown to be active in a handful of case reports of PD-L1-positive UCP,222,223 and a formal assessment of the activity of ICI against UCP is warranted. Additionally, ICIs have been used with success in the treatment of TMB-high UCOGCP. 224
Overall, patients with UCP have inferior survival outcomes when compared to those with PDAC,25,111 but there seems to be no survival difference in the subgroup of patients with resected disease. 25 Also, different subtypes of UCP do not necessarily share the same prognosis. Patients with pleomorphic (anaplastic) undifferentiated carcinomas are more likely to forego surgical resection and have a worse prognosis.26,27 Contrarily, UCOGCP is associated with improved survival,25,26 with 5-year survival rates as high as 59% after resection. 29 It is possible that these higher survival figures are related to improved immune response secondary to the presence of the osteoclast-line multinucleated cells in the tumor microenvironment. 30 Additionally, some, 30 but not all studies, 29 suggest patients with pure UCOGCP have a better prognosis when compared to those with a PDAC component. In the localized setting, R0 resection and negative lymph nodes are harbingers of improved survival.26,27 In the locally advanced and unresectable setting, age ⩾65 years, ECOG PS ⩾2, and CRP ⩾10 mg/L are associated with inferior outcomes. 225
Primary pancreatic signet ring (poorly cohesive) cell carcinoma
Primary pancreatic signet ring (poorly cohesive) cell carcinomas (PPSRCCs) are defined by the current WHO Digestive System Tumor Classification as a histological subtype of PDAC in which at least 80% of the tumor consists of individually arrayed and poorly cohesive cells, often with intracellular mucin vacuoles peripherally displacing the nuclei. 19 This is a rare clinical entity, representing <1% of all malignant pancreatic tumors.33,34 There have been fewer than 15 cases reported in the literature so far, and even though most cases have been reported after 2010, 226 epidemiological studies suggest a decrease in the incidence of this histological subtype over the past two decades. 33
The median age at diagnosis of PPSRCC is 68 years, with a slight male predominance. 32 PPSRCCs usually arise in the pancreatic head (50–90%).32,34 Most patients have metastases at the clinical onset (62–69%)32,33 and follow an aggressive clinical course. 227 The most frequent symptoms are abdominal pain, jaundice, and weight loss.118,226–232 Tumor markers, including CEA and CA 19-9, are variably elevated. 226 There is no clear radiological feature that suggests the diagnosis of PPSRCC over PDAC, 233 however, a few cases of PPSRCC causing diffuse pancreatic enlargement have been reported.230,232 As in gastric signet ring carcinoma, 234 PET-CT seems to have limited capacity to identify PPSRCC. 227 Resected PPSRCCs display a very high rate of vascular and perineural invasion, along with regional lymph node metastases.32,34 In the metastatic setting, the lungs are usually the first site of metastasis, but widespread disease dissemination often follows shortly. 34
In addition to the individually arrayed poorly cohesive cells, 19 there is usually a variable amount of extracellular mucin. The carcinoma cells are positive for mucins on alcian blue/periodic acid–Schiff staining, cytokeratin AE1/3, CAM5.2, CK7, CK8, CK18, CK19, CEA, epithelial membrane antigen (EMA), p53, Ki-67, MUC1, MUC5AC, and MUC6.34,231 PPSRCC is a challenging pathological entity, as cells with signet ring appearance have been reported in a broad array of benign and malignant processes. 235 Especially in the setting of FNA, potential malignant mimickers of PPSRCC include other primary pancreatic tumors with clear cell features, mixed adenoneuroendocrine carcinomas, and pancreatic colloid carcinomas. FNA samples have been reported to show abundant inflammatory cells, potentially delaying the diagnosis.227,228,230 It is important to highlight that >96% of all signet ring carcinomas arise in the stomach, 230 with the remaining arising in other sites, such as the colon, rectum, gallbladder, breast, prostate, and pancreas. Therefore, before establishing the diagnosis of PPSRCC, one should exclude the possibility of metastatic signet ring cell carcinoma secondarily involving the pancreas. 19
Recent data suggest the molecular background of PPSRCC is similar to that of PDAC. In the only series so far to assess the molecular alterations found in pancreatic tumors consisting of ⩾50% of signet ring cells, mutations in KRAS (70%), TP53 (55%), CDKN2A (20%), and SMAD4 (25%) were found in slightly lower rates than those seen in PDACs. 34 dMMR/MSI-H has been found in about 5% of PPSRCC and the median TMB is 7.2 mut/Mb (ranging from 1.0 to 19.4). 34 Additionally, somatic mutations in CDH1, found in 11% of gastric signet ring cell carcinomas, 236 were not found in PPSRCCs. 34 These data indicate that PPSRCC is not derived from ectopic gastric tissue within the pancreas, but rather from clonal evolution of a carcinoma arising in the pancreatic tissue.
In the localized setting, surgery is indicated for all patients with resectable non-metastatic PPSRCC.32,33 There is barely any data to guide decisions regarding adjuvant therapy. However, across all clinical stages, chemotherapy has been associated with improved survival. 33 Nakamura et al. 228 treated one patient with a multi-visceral resection followed by adjuvant S-1 for a year with long-term disease-free survival. Similarly, Radojkovic et al. 118 treated a patient with borderline resectable PPSRCC with neoadjuvant single-agent gemcitabine, achieving a partial response that allowed a curative-intent surgery and a clinically significant disease-free interval. In the metastatic setting, there is no clinical data regarding the activity of chemotherapy, as patients usually follow an aggressive clinical course that precludes the administration of systemic therapy. Given the frequent presence of a component of ordinary PDAC concomitantly with signet ring cell carcinoma, the similarity in the molecular profiles of PPSRCC and PDAC, and the fact that single-agent gemcitabine seems to be active against PPSRCC, we suggest that pending further data patients with PPSRCC should be treated similarly to PDAC, including the chemotherapy regimens (FOLFIRINOX or gemcitabine plus nab-paclitaxel). Importantly, 15% of PPSRCCs seem to harbor potentially actionable molecular alterations, such as dMMR/MSI-H, RET rearrangements, or mutations in PTEN or EGFR (the latter in the absence of KRAS mutation). 34 Therefore, molecular analyses might help identify patients with PPSRCC who might benefit from molecularly tailored treatments.
PPSRCC are aggressive neoplasms with inferior relapse-free survival and OS when compared to PDAC.32–34 The presence of metastatic disease is associated with a dismal prognosis and median OS in this setting in only 3 months. 32 Indeed, PPSRCC portends the worst prognosis among all gastroenteropancreatic signet ring cell carcinomas. 237 Also, patients whose tumors consist of ⩾80% of signet ring cells have inferior survival, as those with SMAD4-mutated tumors. 34
Primary pancreatic hepatoid carcinoma
Tumors arising outside the liver that resemble hepatocellular carcinoma (HCC) are called hepatoid carcinomas (HCs). 238 HCs and HCCs share similar morphological features and both can produce alpha-fetoprotein (AFP). 37 This rare entity is found throughout the gastrointestinal tract, with most tumors arising in the stomach. 239 Less than 40 cases of primary pancreatic hepatoid carcinoma (PPHC) have been reported, representing roughly 4% of all HCs. 239 The pathogenesis of PPHC is not completely understood. Among the current mechanisms to explain its development are the presence of ectopic hepatic tissue in the pancreas, the pancreatic-to-hepatic transdifferentiation, and the presence of multi-potent/stem cells that differentiate into hepatocyte-like cells given the embryological relatedness of hepatic and pancreatic cells. 35
Patients with PPHC are usually male (70%) and tend to be younger than those with PDAC (median age = 58 years). 35 Contrary to PDAC, it arises most often in the pancreatic body/tail and, at presentation, 36–44% of the patients have metastases, mainly in the liver.35,36 Two-thirds of the patients are symptomatic at presentation and, in a recent review of 39 cases, the most common symptoms were abdominal pain, vomiting, diarrhea, weight loss, and jaundice. 35 Serum CEA and CA 19-9 levels are elevated in less than 30% of the cases and serum AFP levels are elevated in 35–60% of patients.35,36,239 However, it is important to highlight AFP production is nonspecific and can be found in non-hepatoid tumors such as PDACs, pancreatoblastomas, and acinar cell carcinomas. 240 On cross-sectional imaging, PPHCs are well-delineated and arterially hypervascularized, showing venous washout similar to HCC. 233
PPHC is defined as a carcinoma in which at least 50% of the tumor exhibits histological and immunohistochemical evidence of hepatocellular differentiation. 19 These tumors are primarily composed of large polygonal cells with abundant eosinophilic cytoplasm. While some PPHCs may have an associated component of ductal adenocarcinoma, others are likely related to acinar cell carcinomas, which can express hepatocellular markers and produce AFP. It is important to note that while AFP is commonly associated with hepatocellular differentiation, it can also be present in other pancreatic neoplasms such as pancreatoblastomas, acinar, neuroendocrine, and ductal tumors without hepatoid morphological features.241,242 For more reliable confirmation of hepatocellular differentiation, fluorescence in situ hybridization for albumin and immunohistochemistry for arginase are recommended. 19 Importantly, before the diagnosis of PHCC can be established, the possibility of a primary hepatic HCC with metastases to the pancreas has to be excluded. 19
Due to its rarity, the molecular alterations underpinning the development of PPHC are not known. In one case report, tumor genetic sequencing revealed BAP1 and NOTCH1 mutations. No genetic alterations were identified in other 539 genes, including KRAS. 38 In one other case of PPHC, both CDKN2A and CDKN2B were mutated. 37 Also, in the case of PPHC with prominent lymphoid infiltration (therefore called HC of the pancreas with lymphoid stroma), neither KRAS mutations nor dMMR/MSI-H were detected. 243 Last, in a recent analysis that included HCs from other gastrointestinal tract sites, the mutational profile of HC has been demonstrated to be dependent on the primary tumor site, recapitulating the genomic alterations commonly observed in the most common malignant epithelial counterpart. 244 This way, even though KRAS mutations have not been described in PPHC, it is possible that many PPHCs share molecular alterations with PDAC, especially those with a more aggressive clinical course.
Long-term survival for patients with localized PPHC depends on curative-intent surgery. 35 Indeed, even in the setting of limited metastatic disease, there seems to be a role for surgery after a period of chemotherapy in properly selected patients.119,120 Given its rarity, there is very little information to guide post-operative treatment decisions. A few case reports describe the use of gemcitabine, gemcitabine plus carboplatin, FOLFIRINOX, and cisplatin plus irinotecan with disease-free survival times of 36, 22, 14+, and 12 months, respectively,245–248 Nonetheless, in cases of lymph node-negative disease, long-term disease-free survival has been reported with observation38,121,122 and it is currently unknown whether all patients with localized PPHC should receive adjuvant chemotherapy. Given the worse prognosis of patients with lymph node metastases, 249 we believe it is reasonable to recommend adjuvant chemotherapy to patients with lymph node-positive disease. Adjuvant sorafenib has been tried in the localized setting, 250 but long-term outcomes are not known.
In the metastatic setting, little is known about the efficacy of systemic treatments against PPHCs. In a case report, single-agent gemcitabine did not show significant activity. 251 Contrarily, FOLFIRINOX has been shown to be active in the metastatic setting. 119 In a case report by Ma et al., 119 a patient with synchronic hepatic metastasis from PPHC experienced a complete pathological response in the liver lesion and a partial response in the primary tumor. Lastly, sorafenib has been used in two patients. One of them was treated for 7 months, with an OS of 12 months. 123 The other had a brief clinical benefit, dying 4 months after the diagnosis. 124 Lastly, PHCC with neuroendocrine features might be sensitive to capecitabine plus temozolomide. 252 Therefore, given the relative success of FOLFIRINOX in the adjuvant and the metastatic setting and the potential role played by BAP1 in the homologous recombination DNA damage repair pathway, FOLFIRINOX seems like a reasonable option for patients with good performance status. 253
Pancreatic medullary carcinoma
Pancreatic medullary carcinoma (PMC) is an extremely rare subtype of pancreatic ductal carcinoma. It was first described by Goggins et al. in 1998 and since then less than 30 cases have been reported.40,42
Clinically, PMCs are frequently large tumors (median size = 5.0 cm) located predominantly in the pancreatic body/tail. 40 Median age at presentation is 67 years and PMCs affect men with a slightly higher frequency than women. 40 Metastases at diagnosis were present in <10% of reported cases.40,126 At clinical presentation, patients might be asymptomatic42,126 or present nonspecific symptoms, such as epigastric pain, nausea, tiredness, and early satiety.40,254–256 A family history of cancer is very common among patients with PMC, 39 and PMC has been described in the setting of Lynch syndrome. 254 Normal levels of CA 19-9 have been described in PMC.40,42 Since carcinomas with medullary features are much more common in the ampulla or duodenum, it is important to exclude that the tumor originated in the adjacent gastrointestinal tract. 19
PMCs are poorly differentiated carcinomas with limited gland formation, pushing border, and syncytial growth pattern, often with a rich lymphocyte infiltration. 19 Molecularly, PMCs commonly lack KRAS mutations. 39 Additionally, dMMR/MSI-H is more common in MCP than in PDAC. In a recent compilation of all cases reported in the literature so far, 25% of PMCs showed dMMR/MSI-H, 40 mostly secondary to loss of MLH1 expression. 39 Also, somatic mutations in POLE leading to a hypermutated phenotype have also been reported. 255
Surgery is the cornerstone of treatment in the localized setting. 125 Additionally, in patients with oligometastatic disease, radical resection can achieve prolonged survival and should be considered. 126 Currently, the role of neoadjuvant and adjuvant chemotherapy for PMC is unknown. Both S-1 and FOLFIRINOX have been used in the adjuvant setting, with disease-free survival times of 29+ and 7+ months, respectively.40,42 However, patients with long-term survival after upfront surgery without adjuvant chemotherapy have been reported and it is unclear whether regional lymph node metastasis represents a true prognostic factor, as long-term survival in this setting has been repeatedly reported.255,257 In one of the authors’ personal experiences (VHFdJ), one patient with dMMR/MSI-H PMC treated with neoadjuvant mFOLFIRINOX achieved a complete pathological response in the setting of regional lymph node metastasis (case report currently being written). This suggests fluoropyrimidine-based chemotherapy (especially FOLFIRINOX) might be active in the treatment of PMC. In one case report, gemcitabine plus nab-paclitaxel was given to a patient with metastatic disease for 5 months before clinical deterioration. 256 Also, dMMR/MSI-H pancreatic cancer, including PMCs, are sensitive to ICIs,127,128 ratifying the need to investigate dMMR/MSI-H status in these tumors. Additionally, patients with hypermutated tumors due to pathogenic POLE mutations in the DNA binding or catalytic sites of the exonuclease domain have been shown to benefit from ICIs. 129
Other pancreatic epithelial malignancies
Pancreatic acinar cell carcinoma
Pancreatic acinar cell carcinoma (PACC), along with mixed acinar carcinomas, acinar cystic lesions, and pancreatoblastomas, belongs to a group of pancreatic neoplasms that show acinar differentiation. 258 It represents 1–2% of all pancreatic neoplasms in adults and 15% in children. 54 Despite its rarity, the results of epidemiological studies indicate the incidence of PACC has increased at a much faster pace than that of PDAC over the past two decades.59,259 No specific risk factors for the development of PACC have been established, but tobacco smoking has emerged as a potential risk factor since recent studies have identified a smoking mutational signature in two-thirds of PACCs. 260 Additionally, PACC has rarely been described in the setting of Lynch syndrome, familial adenomatous polyposis, and Carney syndrome.54,258,261
PACC is more common among men (sex ratio = 2:1 to 4:1) and affects younger patients when compared to PDAC (median age = 54–67 years old).43–49 Approximately 50% of patients have metastases at presentation, usually in the liver and lymph nodes. 258 The following are the most frequent symptoms: abdominal pain, back pain, bloating, weight loss, and nausea/vomiting.47–49,130,136,258,262 Even though most PACCs arise in the pancreatic head (44–76%), 45 due to its expansible (non-infiltrative) growth pattern, jaundice is not a common symptom (<25% of patients).47–49,136,262 AFP levels are increased in less than 10% of patients, 49 especially in pediatric cases 261 and CEA and CA 19-9 levels can also be elevated, but to a lesser extent to that seen in PDAC.49,59,131 Lipase levels are increased in roughly half of the patients.46,263 Less than 10% of the patients present with lipase hypersecretion syndrome, with lipase levels exceeding 10,000 U/L. These patients often present multiple foci of subcutaneous fact necrosis (simulating erythema nodosum), polyarthralgia (due to fat necrosis in cancellous bone), and peripheral eosinophilia – Schmid’s triad.264,265
On cross-sectional imaging, PACCs generally present as large, solid, and well-circumscribed tumors.50–53 In contrast with PDAC, bile duct dilatation is uncommon, even when tumors arise in the pancreatic head.50,51 In the arterial phase, PACCs often display more contrast enhancement compared to PDAC, but less than that of well-differentiated neuroendocrine neoplasms (WD-NEN). 266 Calcifications and central necrosis have also been described with a higher frequency than in PDAC.50,51,53
PACCs typically exhibit well-defined borders, being partially encapsulated, solid, and often substantial in size (with an average diameter ranging from 8 to 10 cm). 54 These tumors may present hemorrhage and necrosis and may exhibit different architectural patterns. 54 The acinar pattern resembles normal acinar structures, sometimes with tiny lumina, and cells are arranged in a monolayer with basally located nuclei. The glandular pattern features acinar structures with dilated lumina. The trabecular pattern presents as ribbons of cells resembling pancreatic neuroendocrine tumors (PanNETs), while the solid pattern consists of large sheets of cells without lumina, resembling PanNETs as well. The most common patterns are acinar and solid, often found in combination within individual tumors. 54
Immunohistochemistry plays a crucial role in confirming acinar cell differentiation. Antibodies targeting trypsin, chymotrypsin, lipase, and amylase show varying sensitivity, with the antibody against the COOH-terminal portion of the BCL10 protein (clone 331.3), 267 which recognizes the COOH-terminal portion of pancreatic carboxyl ester lipase, being highly specific and sensitive. Amylase expression is rare, and lipase antibodies have low sensitivity, while trypsin, chymotrypsin, and BCL10 antibodies are more sensitive. 54 The simultaneous use of two of these antibodies allows for the detection of nearly all PACCs. Markers typically associated with HCCs, such as AFP, Hep Par-1, glypican-3, and albumin mRNA, can also be detected in PACCs. 268 Finally, scattered neuroendocrine cells positive for chromogranin A and/or synaptophysin may also be observed. 54
The molecular landscape of PACC is very distinct from that of PDAC. In PACC, mutations in KRAS are rare (<15%).23,261,269 Moreover, mutations in TP53 (12–23%), CDKN2A (12–14%), and SMAD4 (14–26%) are significantly less common than in PDAC.56,260,261,270 No single gene is mutated in more than 30% of PACCs, 270 with mutations in FAT4, RB, BRAF, GNAS, PRKAR1A, NF1, and JAK1 been reported in multiple studies.56,260,269,270 The APC gene is mutated in 10–20% of PACCs 260 ; however, APC loss and methylation seem to be even more common, being present in 48% and 56%, respectively. 271 Interestingly, in mouse embryos, APC mutations lead to pancreatomegaly secondary to selective proliferation of acinar cells. 261
From the structural perspective, PACCs often display highly unstable genomes, with localized and broad-ranged chromosomal gains and losses. 260 Recent analyses suggest two distinct molecular phenomena are associated with these structural alterations. Defects in the homologous recombination repair (HRR) pathway are found in 45–55% of PACCs.55,56 Indeed, PACC is the tumor most frequently associated with inherited defects in HRR genes (mainly BRCA2 and ATM), with 37% of patients with PACC harboring germline mutations in such genes, especially in cases of pure PACC. 55 The importance of the HRR pathway is highlighted by the fact that in the same series, all patients with germline HRR gene mutation had biallelic gene inactivation in tumor tissue.55,269 Additionally, rearrangements in RAF1 (19%), BRAF (14–20%), and RET (8%) occur in at least one-third of PACCs23,57,58 and are more frequent in pure PACCs.56,57 These rearrangements involving the mitogen-activated protein kinase (MAPK) pathway seem to be mutually exclusive, 57 and there seems to be no overlap between these rearrangements and HRR defects.55,56 Despite the relatively low median TMB of PACC (3–4 mutations/Mb),55,272 7–8% have TMB >10 mutations/Mb.23,273 Moreover, 8.2% (0–14%) of PACCs display dMMR/MSI-H.261,273–276
Surgical resection is one of the most important prognostic factors for patients with localized PACC.49,130,131 Furthermore, case reports and small case series suggest patients might benefit from surgery even in the setting of metastases.132–134 The role of adjuvant chemotherapy in PACC is more controversial. While some small studies suggest a positive effect of chemotherapy after resection,48,49 others have failed to show improved survival for patients receiving adjuvant chemotherapy.43,135,136 Larger and more recent population-based studies have suggested adjuvant chemotherapy is associated with improved survival, especially for patients with lymph node-positive disease.137,138 Two facts might explain the disputable activity of chemotherapy in the adjuvant setting. Molecular data suggest that up to 14% of PACC have mismatch repair deficiency/MSI-H 274 and, at least in colon and gastric cancer, adjuvant chemotherapy with 5-fluorouracil-based chemotherapy has failed to improve survival in this subgroup, especially for lymph node-negative disease.136,277,278 Additionally, in the series that report the chemotherapy backbone used in the adjuvant setting, most patients received gemcitabine-based chemotherapy, and at least in the metastatic setting, 5-fluorouracil-based chemotherapy (especially when combined with oxaliplatin and/or irinotecan) seems to be more active than gemcitabine-based combinations. Therefore, given that recurrence rates after resection as high as 72% have been described 130 and the increased activity of 5-fluorouracil-based polychemotherapy regimens in the metastatic setting, we suggest fit patients with resected PACC should receive adjuvant chemotherapy with FOLFIRINOX for 6 months, especially those with lymph node-positive disease.
Conversion strategies have been successfully used in the setting of unresectable disease, either with chemotherapy or chemoradiotherapy, and should be considered for all patients with locally advanced PACC.44,135,262 Last, limited population-based data suggest adjuvant chemoradiotherapy might be associated with improved survival for patients with PACC. 139 This is somewhat surprising given its lack of survival benefit for PDAC, 279 the lesser frequency of positive margins after pancreatectomy for PACC, 43 and the less infiltrative growth pattern of PACC. 136 Besides, similarly to PDAC, distant metastasis is the single main pattern of disease relapse in PACC.131,135 Therefore, routine use of adjuvant chemoradiotherapy for patients with resected PACC does not seem justified.
Initial experience with systemic chemotherapy for advanced PACC was disappointing, with objective response rates under 10%. 130 However, recent studies suggest newer chemotherapy regimens are more active against PACC. 46 Particularly, studies have shown that 5-fluorouracil-based combinations demonstrate improved efficacy when compared with gemcitabine-based regimens.140,141 Importantly, improved efficacy of irinotecan- and oxaliplatin-based regimens has been shown in single-center and multi-institutional analyses.23,46,115,141,263,280 The reasons for this apparent sensitivity to 5-fluorouracil, irinotecan, and oxaliplatin are not entirely understood. Limited data suggest pancreatic carcinomas with RAF fusions are more sensitive to 5-fluorouracil-based chemotherapy. 144 Also, the high frequency of APC/b-catenin pathway alterations in PACC (nearly 50%) 271 might explain the sensitivity to chemotherapy regimens used in colorectal cancer, a disease that often harbors alterations in this molecular pathway. 281 Finally, it has been shown that 55% of PACC have defects in the HRR pathway, which is associated with increased efficacy of platinum-based compounds.55,282 Interestingly, limited evidence suggests FOLFIRINOX might be more effective than gemcitabine plus nab-paclitaxel even in the absence of HRR defects. 23
As previously described, two main types of genetic events seem to drive the initiation and progression of PACC: mutations in HRR genes and fusions involving RAF1, BRAF, and RET. 55 Multiple case reports have demonstrated the activity of poly(ADP-ribose) polymerase inhibitors in patients with PACC and BRCA1/2 germline mutations.142,143 Indeed, given the very high frequency of HRR defects in PACC, one study is currently testing the activity of olaparib in treatment-refractory PACC irrespective of the homologous recombination status (NCT05286827). The combination of BRAF and MEK inhibitors is active against PACCs with BRAF V600E mutations.269,283–285 MEK inhibitors have shown clinical activity against RAF fusion-positive PACC, 144 with two patients (among five) experiencing partial radiological response. This is particularly important since patients with BRAF fusion-positive PACC usually have a shorter time to treatment failure. 23 Additionally, clinical activity of targeted therapy against tumors harboring fusions involving other genes, such as RET (selpercatinib), 145 ALK (alectinib), 146 and NTRK1 (larotrectinib), 147 have been described. Importantly, it has been shown that up to 14% of PACCs have mismatch repair deficiency/MSI-H and anti-PD-1 monoclonal antibodies should be considered for patients with advanced dMMR/MSI-H PACC based on the data on PDAC.127,128,286 In contrast to other tumors, dMMR/MSI-H PACCs do not display prominent lymphoid infiltration, 274 possibly suggesting that the combination of anti-PD-1 and anti-CTLA4 monoclonal antibodies is needed. Finally, despite the overall low TMB of PACC, in one case report, a patient with TMB-high advanced PACC was successfully treated with the anti-PD-1 monoclonal antibody toripalimab in combination with chemotherapy. 287
Pure PACC and mixed acinar-neuroendocrine carcinomas share a similar prognosis,288,289 and after controlling for other factors, the prognosis of PACC is better than that of PDAC. 59 The co-occurrence of TP53 mutation and TP53 loss has been associated with worse survival 290 and dMMR/MSI-H PACC does not seem to have improved outcomes. 274 While there are no established prognostic factors in the metastatic setting, advanced age, presence of lymph node involvement (especially N2 disease), elevated CA 19-9 levels and perineural invasion are associated with poor prognosis for patients with localized disease.135,136,289,291
Adult pancreatoblastoma
Pancreatoblastomas are rare primary malignant neoplasms of the pancreas that affect mainly children (median age = 4–5 years). 64 However, the age of presentation of pancreatoblastoma follows a bimodal distribution, with a second peak at roughly 40 years of age. 65 Indeed, one-third of all reported cases of pancreatoblastoma were diagnosed in adults. 292 So far, approximately 74 cases of adult pancreatoblastoma (AP; defined as age ⩾19 years) 149 have been reported, 61 and AP represent 0.5% of all malignant exocrine pancreatic tumors. 60
The median age at presentation of AP is 37–41 years.60–63 There seems to be no sex predilection and most tumors arise in the pancreatic head.60,61 Tumors are usually large (median size = 8 cm) and up to one-third of patients have metastatic disease at the initial staging, typically in the liver.60,61,63 Most patients are symptomatic at the disease presentation, with the most frequent symptoms being abdominal pain, nausea and vomiting, weight loss, jaundice, and diarrhea.60,62,65 In contrast to pediatric pancreatoblastoma, adult patients rarely have increased levels of AFP (6%), and similarly, CA 19-9 levels are seldom increased in AP (6%).60,63,66,293 In line with the role of APC in the Wnt-signaling pathway, a few cases of APs have been described in the setting of Familial Adenomatous Polyposis. 294 On cross-sectional imaging, APs appear as irregular hypovascular masses that grow outwards from the pancreas. 295 Upon contrast injection, APs usually present a progressive enhancement pattern, 295 but an early enhancement pattern has also been described. 296
Most of these tumors are large solid solitary masses, at least partially exhibiting well-defined or encapsulated characteristics. 297 Histologically, pancreatoblastomas exhibit highly cellular lobules separated by fibrous bands, creating a distinct geographical or lymphoid-follicle, multiphasic low-power appearance. 64 The neoplastic cells within the lobules typically demonstrate an organoid arrangement resembling acinar cell carcinomas, appearing as acinar, solid, or trabecular formations polarized around small lumina, with nuclei typically displaying modest nuclear atypia. The presence of acinar differentiation is supported by periodic acid-Schiff with diastase (PASD)-positive cytoplasmic granules and positive immunohistochemical labeling for pancreatic enzymes (trypsin, chymotrypsin, and lipase) and BCL10 (clone 331.1). 64
A defining feature of pancreatoblastoma is the presence of squamoid nests, crucial for establishing the diagnosis. 298 These nests comprise distinctive cells with eosinophilic to clear cytoplasm, forming islands or whorled nests with a squamous appearance, sometimes exhibiting overt keratinization. The nuclei of these cells are larger and more oval-shaped than those of surrounding cells and lack prominent nucleoli or mitoses. Occasionally, nuclear clearing due to intranuclear accumulation of biotin may be observed. 299 Squamoid nests also stain positive for EMA. In addition to the acinar component and squamoid nests, a neuroendocrine component can be detected using specific immunohistochemical stains (chromogranin A, synaptophysin). In adults, squamoid nests and cellular fibroma might be more subtle, making the distinction between PACC and AP difficult.64,258 In doubtful cases, it is suggested to favor the diagnosis of pancreatoblastoma in children and PACC in adults. 258
Molecular events involving the Wnt-signaling pathway are a hallmark of pancreatoblastoma. Activating mutations in exon 3 of CTNNB1 occur in 90% of pediatric pancreatoblastomas and similar findings have been shown in the adult population. 300 Loss of 11p have been described in a comparable frequency and inactivating mutations in APC have also been reported.65,301 However, CTNNB1 and APC mutations are considered to be mutually exclusive. 66 Also, mutations in KRAS, CDKN2A, and TP53 are typically lacking in pancreatoblastoma,61,302 and SMAD4 mutations have occasionally been described. 270 ALK, CTLA4, PDCD1, CALML3, and KLK4 seem to have higher gene expression in comparison to acinar cell carcinomas. 302 Other reports describe MCL1 amplifications (15%) and alterations in FGRF signaling pathways are potentially targetable molecular events.150,303 Pancreatoblastomas usually present a low TMB and, so far, no case of dMMR/MSI-H AP has been reported.65,66,270
Surgical resection is the cornerstone of curative treatment,60,148,149 and at least in pediatric cases, R0 resections seem to be associated with improved prognosis. 304 In pediatric pancreatoblastoma, long-term survival with resection of metastatic disease following chemotherapy has been reported. 305 However, in AP, most data suggest patients experience short-lived benefits from resection of metastatic disease.150,306 Whenever an upfront R0 resection is not feasible, especially in cases of large and aggressive tumors, neoadjuvant chemotherapy can be recommended.304,307 In children, pancreatoblastoma is considered to be a chemosensitive tumor and the chemotherapy regimen most frequently used is doxorubicin plus cisplatin (PLADO regimen). 304 In children, response rates of up to 73% have been described. 305 However, this chemotherapy regimen has been shown to be rather ineffective and toxic in adults,115,150 and more recently, FOLFIRINOX has been shown to be active both in the neoadjuvant and metastatic settings.66,150,151 Also, limited data suggest gemcitabine-based chemotherapy has little activity against AP. 115 After complete resection of an AP, there is no robust data regarding the role of adjuvant chemotherapy. While chemotherapy is commonly indicated for patients with lymph node-positive pediatric pancreatoblastoma, 304 the available evidence suggests there is no benefit of adjuvant chemotherapy for AP. 60 However, adult patients with lymph node-positive disease seem to experience an aggressive clinical course, and given the recently reported activity of FOLFIRINOX in this setting, we believe it is reasonable to recommend adjuvant FOLFIRINOX for these patients. Limited data suggest adjuvant radiotherapy is not beneficial against AP. However, radiotherapy can be used to palliate symptoms in the metastatic setting, and, based on data from pediatric pancreatoblastoma, it can be considered in cases of positive surgical margins.60,304 In the pediatric setting, high-dose chemotherapy with peripheral stem cell rescue has been employed in selected cases, 304 and mixed results have been described in adults.150,308 Therefore, we believe this could be a treatment option in very selected cases of disease progressing to first-line chemotherapy.
The upregulation of immune checkpoint-related genes suggests immunotherapy might play a role in the treatment of AP. 302 However, a recent report describes the use of ICIs in two patients with AP with disappointing results. 150 Likewise, the anti-FGFR (fibroblast growth factor receptor) agent nintedanib has been used in a patient with AP harboring an FGRF2-INA fusion with no significant clinical benefit. 150
The prognosis of AP is worse than its pediatric counterpart.61,65 However, pancreatoblastomas in adults usually follow a less aggressive clinical course when compared to PDAC. 66 Advanced age, presence of lymph node metastasis, and unresectable disease are considered to be poor prognostic factors for patients with AP.60,65
Invasive intraductal papillary mucinous neoplasms
Pancreatic cystic lesions (PCL) are very common in the general population. In cross-sectional imaging studies, the prevalence of PCL has been estimated to be between 2% to 15%, increasing to up to 50% in autopsy series. 309 While in the past pseudocysts accounted for a large number of pancreatic cysts, currently up to 95% of PCLs are pancreatic cystic neoplasms (PCN). 309 Intraductal papillary mucinous neoplasm (IPMN) is the most clinically relevant variety of PCN, representing 21–38% of all resected PCNs. 310 It is defined by the current WHO Digestive System Tumor Classification as grossly visible (typically >5 mm) intraductal epithelial neoplasm of mucin-producing cells, arising in the main pancreatic duct and/or its branches. 311 Importantly, the incidence of IPMN is rising considerably in the West as a consequence of increased awareness of this clinical entity, albeit one cannot exclude the possibility of increased exposure to still non-identified risk factors.310,312
IPMNs affect men slightly more often than women and frequently occur in the sixth to seventh decades of life. 313 Interestingly, patients with invasive IPMNs are 3–5 years older than those with non-invasive IPMNs, suggesting a time lag of several years before non-invasive IPMNs can progress to overt malignant tumors.311,313 With the widespread use of non-invasive abdominal imaging techniques, IPMNs are often incidentally detected when imaging is performed for unrelated conditions. 71 When symptomatic, IPMNs can cause abdominal pain, acute pancreatitis (especially if MD-IPMN), jaundice, weight loss, steatorrhea, and worsening of glycemic control.314–316 Importantly, the risk of invasive carcinoma is higher for symptomatic patients and there is a positive correlation between the duration of symptoms and the chance of malignancy in IPMNs.71,314,317
Patients with IPMN can develop pancreatic cancer in the two following scenarios: histological transformation of IPMN to invasive cancer and concomitant PDAC that can co-occur far from or in the vicinity of an IPMN. 318 In the latter case, it might be difficult to distinguish invasive IPMN from concomitant PDAC. 319 According to the classification proposed by the Japanese Pancreas Society, invasive IPMN is thought to be derived from IPMN based on radiological, macroscopic, and microscopic findings, with a histological transition between the two components. 320 Conversely, concomitant PDAC does not have clear radiological or pathological relatedness to the adjacent IPMN or a transition zone. This distinction is clinically important since the prognosis of invasive IPMN is better than that of concomitant PDAC. 318 Additional pieces of information that can help differentiate invasive IPMN from concomitant PDAC are the presence of colloid carcinoma in association with intestinal-type IPMN and GNAS mutation in the invasive component, which favors the diagnosis of invasive IPMN. 319 Conversely, the absence of mural nodule in the IPMN favors the diagnosis of concomitant PDAC and it is important to bear in mind that IPMN size and pancreatic duct dilatation are associated with the risk of invasive IPMN, but not of concomitant PDAC. 321
Invasive carcinoma associated with IPMNs can manifest as two distinct types. 311 Colloid carcinoma, characterized by infiltrating epithelial elements separated by abundant stromal mucin, typically arising alongside intestinal-type IPMNs. On the other hand, tubular (ductal) adenocarcinoma, resembling conventional PDAC morphologically, is linked with both pancreatobiliary-type and gastric-type IPMNs. Ductal markers like CK7, CK19, CA 19-9, and CEA are commonly expressed in most IPMNs. The expression pattern of mucins and CDX2, 322 a marker of intestinal differentiation, aids in distinguishing the morphological subtypes, 323 although overlapping expression patterns are common. 324
Activating mutations in KRAS and GNAS are the most frequent and earlier genetic events reported in IPMNs. 73 Mutations in KRAS occur in 50–80% of IPMNs. 74 GNAS encodes the stimulatory a-subunit of the G-protein-coupled receptor, which activates Protein Kinase A through increased levels of cyclic adenosine monophosphate. Activating mutations in the codon 201 hotspot of GNAS are reported in 40–70% of all IPMNs, and are more common in intestinal-type IPMN. 319 GNAS mutations seem to be less frequently found in invasive IPMN 325 and colloid-type invasive IPMN seems to harbor GNAS mutations at a much higher rate than tubular-type invasive IPMN (89% versus 32%). 326 Loss-of-function mutations in RNF43 (a negative regulator of the Wnt-signaling pathway) have been reported in 10–75% of IPMNs and mutations in other cancer-related genes have also been described at a low prevalence, such as BRAF, PIK3CA, STK11, and PTEN. 74 Late molecular events present in high-grade IPMN involve loss-of-function changes in tumor suppressor genes such as TP53, CDKN2A, and SMAD4, 327 with alterations in the latter gene reported exclusively in invasive IPMN. 74
Both low-grade and high-grade IPMNs have a low median TMB (around 1 mutation/Mb).73,328 However, IPMN-related cancers harbor a significantly higher frequency of dMMR/MSI-H (7%) than non-IPMN-related PDACs (1%) and loss of MMR protein expression is present in both IPMN and invasive IPMN. 329 A recent systematic review of 34 studies with 8323 patients with pancreatic cancer demonstrated a very low prevalence of dMMR/MSI-H in PDAC (up to 2%). However, medullary and mucinous/colloid histology showed a strong association with MSI/dMMR (p < 0.01). 41 Therefore, dMMR–MSI-H status should be determined as part of routine analysis in IPMN-associated carcinomas, as well as in mucinous/colloid and medullary carcinomas of the pancreas.
Invasive IPMNs are generally diagnosed postoperatively after pathological assessment of the surgical specimen. Indeed, in opposition to PDAC, more than 90% of the patients with invasive IPMN have their disease diagnosed in the localized setting. 69 Currently, there is no RCT assessing the role of adjuvant chemotherapy or radiotherapy for resected invasive IPMN. All available information stems from population-based studies and retrospective uni- or multi-institutional cohorts. While multiple analyses have failed to identify a survival benefit in favor of adjuvant chemotherapy for invasive IPMN,75,330,331 others have found adjuvant chemotherapy is associated with improvements in survival only for selected subgroups of patients, such as those with lymph node-positive disease,77,152–155 tubular adenocarcinomas, 152 higher disease stage (⩾stage II), 77 and poorly differentiated tumors. 77 Indeed, very limited data suggest unselected patients benefit from adjuvant chemotherapy after resection of invasive IPMN. 332 In a recent systematic review and meta-analysis, adjuvant chemotherapy was associated with improved survival only for patients with lymph node-positive or stage ⩾II disease. 333 As a result, we believe patients with lymph node-positive disease, especially of tubular histology (given its worse prognosis), should receive adjuvant chemotherapy. One caveat of these analyses is that most patients given adjuvant therapy were treated with single-agent chemotherapy (especially gemcitabine) 333 and the use of the more active polychemotherapy regimens might have led to more substantial survival improvements. Pending further data specifically for invasive IPMN, we find it reasonable to extrapolate the data from PDAC and recommend the use of newer multiagent regimens, such as FOLFIRINOX, in the adjuvant setting.
Also, in these studies, a variable number of patients received adjuvant radiotherapy 333 and it is currently not known whether adjuvant radiotherapy is beneficial in this setting. While limited data from population-based studies suggest adjuvant radiotherapy is associated with improved survival for patients with cT3/4 or lymph node-positive disease,77,156,157 relapses of invasive IPMN usually occur at distant sites, such as the liver and lungs.334,335 Therefore, we believe the routine use of adjuvant radiotherapy is not warranted.
In the setting of metastatic or relapsed invasive IPMN, there is very limited data on the activity of chemotherapy. In a small cohort from Italy, 13 patients with metastatic invasive IPMN were treated with either single-agent gemcitabine or gemcitabine plus oxaliplatin. The ORR was 15% and the DCR was 61%. 158 Median PFS and OS were 9.6 and 15.4 months, respectively. In a cohort study from Japan, 12 patients with relapsed invasive IPMN received single-agent gemcitabine. 159 No patient experienced partial response and the DCR was 58%. This time, the median PFS and OS were only 2.8 and 9.4 months, respectively. Last, another Japanese study evaluated the activity of single-agent gemcitabine in 14 patients with unresectable invasive IPMN. The ORR was 14% and the DCR was 79%. Median PFS and OS were 8.0 and 24.0 months, respectively. 160 Currently there is no data on the activity of FOLFIRINOX or gemcitabine plus nab-paclitaxel against relapsed or metastatic invasive IPMN. That said, extrapolating the data from clinical trials in PDAC,336,337 we believe both combination chemotherapy regimens are considered reasonable options in this setting.
Most studies suggest invasive IPMN have a better prognosis than PDAC.70,75–77 However, this is not a universal finding after adjusting for other prognostic factors. 72 Also, the difference in prognosis seems to be greater for earlier stages,76,338 and metastatic invasive IPMN have a similar prognosis to that of metastatic PDAC.69,72 Additionally, the prognosis of colloid-type invasive IPMN is better than that of tubular-type invasive IPMN, which has been associated with survival figures similar to those of PDAC.78,79 In the localized setting, T staging, type of invasive component, N staging, histological grade, perineural invasion, and CA 19-9 levels are significant prognostic factors.152,154,330–332 In the metastatic setting, age >60 years, multiple sites of metastasis, and the presence of liver metastasis are associated with inferior survival. 339
Invasive mucinous cystic neoplasm
Mucinous cystic neoplasms (MCNs) are cyst-forming and mucin-producing epithelial neoplasms, 82 which diagnosis requires the presence of an ovarian-like stroma. MCNs represent 8–10% of PCL.82–84 MCN affects females almost exclusively (>90% of patients) and the mean age at diagnosis is between 40 and 60 years.82,83 Patients with invasive MCN are 10–15 older than those with non-invasive MCN.80,81 MCNs almost always arise in the pancreatic body or tail (>98%)82,84 and at presentation nearly half of the patients are symptomatic. 83 The predominant symptoms at presentation are upper abdominal bloating and pain, palpable mass, and weight loss. 81 On cross-sectional imaging, MCNs present as cystic lesions with intralesional septa defining a small number (usually <6) of relatively large cysts (>2 cm). 340 It also lacks communication with the pancreatic ductal system, a characteristic that helps differentiate MCNs from IPMNs. 84
MCNs commonly present as cystic masses with variable thickness of the fibrous wall, occasionally showing calcifications. 82 They can range from 2 to 35 cm in greatest dimension, with a mean size around 6 cm. However, MCNs with invasive carcinoma tend to be larger, with a mean size of approximately 9 cm. 341 Histologically, the cysts of MCNs are lined by epithelium and have underlying ovarian-type stroma. The distinctive ovarian-type stroma found in MCNs consists of densely packed spindle-shaped cells with round or elongated nuclei and sparse cytoplasm. 342 Its presence is essential for the diagnosis of MCNs and can be particularly useful when the epithelial lining is extensively denuded. 82
MCNs are categorized into low-grade and high-grade based on the highest degree of cytoarchitectural atypia in the epithelium. 82 High-grade MCNs display severe atypia, including the formation of papillae with irregular branching and budding. Nuclear features such as stratification with loss of polarity, pleomorphism, and prominent nucleoli are prominent. Additionally, high-grade MCNs often exhibit numerous mitotic figures, indicating increased cellular proliferation and dysplasia. Approximately 15% of MCNs have an associated invasive carcinoma component, typically occurring in large tumors with gross papillary nodules. 82 The invasive component is usually tubular-type adenocarcinoma, although other subtypes have been described. 343 Colloid carcinomas do not occur in MCNs, and the invasive component can be focal, necessitating thorough sampling. 341
Immunohistochemically, the neoplastic epithelial cells in MCNs express various markers including CK7, CK8, CK18, CK19, EMA, CEA, and MUC5AC. 344 While non-invasive MCNs express SMAD4 protein but not EMA (MUC1), invasive carcinomas in MCNs may lose SMAD4 expression and express EMA (MUC1). 345 The ovarian-type stroma, on the other hand, expresses smooth muscle actin (SMA), muscle-specific actin (MSA), desmin, progesterone receptor (PR) (60–90%), and estrogen receptor (ER) (30%).82,346
MCNs have been variably found to harbor activating mutations in KRAS (3–100%), with higher frequency in high-grade lesions.74,86,87 Similar to invasive IPMN, mutations in RNF43 are found in up to 56% of MCNs, also with higher frequency in high-grade and invasive MCNs. 87 Nonetheless, unlike invasive IPMN, GNAS mutations are not found in invasive MCN. 74 CDKN2A loss and TP53 mutations have also been frequently found, almost exclusively in high-grade lesions.74,86 Finally, SMAD4 loss occurs at a high frequency in invasive MCN, but is not described in non-invasive MCN. 347
In a recent systematic review and meta-analysis, invasive carcinoma was found in 16% of resected MCNs. 85 Characteristics associated with increased risk of malignancy include larger tumor size (>5–6.5 cm), presence of mural nodes, irregular thickening of the cyst wall, and increased levels of serum tumor markers (CEA, CA 19-9, and CA 125).82,85 As a result, current guidelines suggest MCNs should be resected in case of lesion size >4 cm, mural nodule or solid mass, positive cytology for high-grade or invasive lesion, or disease-related symptoms. 348
Given its rarity, there is no high-quality data regarding the role of adjuvant chemotherapy for patients with resected invasive MCN. However, most patients with invasive MCN present with lymph node-negative disease, with a lymph node positivity rate ranging from 0% to 34%,68,349–351 and these patients have a much better prognosis than those with invasive IPMN or PDAC, with 10-year OS rates up to 80%.68,88 Also, limited data from population-based studies suggest adjuvant chemotherapy is not associated with improved survival outcomes for patients with resected invasive MCN. 88 Therefore, one cannot routinely recommend adjuvant chemotherapy for patients with invasive MCN.
Again, there is little information to guide treatment decisions for patients with metastatic or relapsed invasive MCN. Werner et al. 161 treated five patients with mucinous cystadenocarcinoma with metastases or vascular invasion with gemcitabine-based chemotherapy, with a mean OS of 11 months (ranging from 4 to 37 months). In one Italian multicenter study, 15 patients with pancreatic mucinous cystadenocarcinoma were given chemotherapy in the first-line setting, mostly (N = 12) gemcitabine-based regimens. 115 Two patients achieved a partial response (one treated with gemcitabine plus erlotinib and the other with gemcitabine plus cisplatin), for an ORR rate of 13.3% and a DCR of 80%. Three among four long-term survivors (OS ⩾ 15 months) received gemcitabine plus cisplatin in first-line. Interestingly, all three patients who received FOLFIRINOX experienced stable disease as the best response, with time to progression ranging from 5 to 7 months. In the second-line setting, one patient (among 12) had a partial response, this time with FOLFOX. Collectively, many case reports suggest interesting activity of gemcitabine against metastatic invasive MCN, especially when combined with a platinum agent.162–165 Additionally, in a few case reports FOLFIRINOX led to tumor shrinkage and prolonged disease control, suggesting this might also be an option.165,352 Therefore, the available clinical data suggest gemcitabine plus a platinum compound (either cisplatin or oxaliplatin) is a reasonable choice in this setting. However, given the limited efficacy of chemotherapy and the dearth of information regarding the activity of more contemporary regimens, such as gemcitabine plus nab-paclitaxel and FOLFIRINOX, we acknowledge such regimens are also acceptable treatment options.
Patients with metastatic invasive MCN have a similar prognosis to that of invasive IPMN. 88 Also, the presence of lymph node metastasis is associated with inferior survival for patients with non-metastatic invasive MCN. 88
Solid pseudopapillary neoplasms of the pancreas
Solid pseudopapillary neoplasms of the pancreas (SPPNPs) are defined by the current WHO Digestive System Tumor Classification as low-grade malignant neoplasms composed of poorly cohesive epithelial cells forming solid and pseudopapillary structures that lack a specific line of pancreatic epithelial differentiation. 99 It represents 10–15% of all PCNs and 1–2% of all pancreatic tumors.95,97 In patients <40 years old, SPPNP represents 30% of all pancreatic tumors. 99 Epidemiological data on this tumor suggest its incidence is increasing, and in fact, most cases have been reported in the past 20 years.89,353
SPPNPs usually affect women of reproductive age. Eighty–ninety percent of all cases arise in women and the mean age at diagnosis is 27–30 years.89–91 At presentation, 37–69% of the patients are symptomatic, and the most frequent symptoms are abdominal or epigastric discomfort/pain, nausea and vomiting, palpable mass, and weight loss.92–97 SPPNP usually arises in the pancreatic body or tail (57–74%).92–97 Interestingly, CA 19-9 is elevated in a minority of patients (0–12%), usually at low levels.92,93,95,97 On imaging, SPPNPs usually are well-delimited and large tumors (mean size ranging from 4 to 9 cm) with a characteristic solid and cystic appearance.90,98 The cystic components represent foci of hemorrhage that occur as the tumor outgrows the blood supply and are better seen in T1-weighted MRI imaging. Calcifications are seen in up to 30% of the cases. 354 Interestingly, even with these distinctive features, the preoperative diagnosis of SPPNP can only be established in 70% of the cases.92,95
Small tumors typically exhibit a denser (solid) consistency and the neoplasm’s wall may harbor calcifications. 99 Infrequently, the tumor extends into the duodenal wall or other neighboring structures. 355 Microscopically, solid and pseudopapillary structures are blended with hemorrhage and pseudocystic changes in varying proportions. 99 The solid tumor component, which may resemble a NEN, comprises poorly cohesive monomorphic cells adhering to hyalinized or myxoid fibrovascular cords. 99 Pseudopapillae form as neoplastic cells detach from the fibrovascular stalks. Although generally well-demarcated, the tumor may infiltrate surrounding pancreatic tissue focally, entrapping acinar cells and islets. Vascular and perineural invasion is rare and metastases generally mirror the primary tumor morphologically, with cells possibly exhibiting more pleomorphism and mitoses. 355 Solid pseudopapillary neoplasms (SPNs) with areas of high-grade malignant transformation are recognized as a histological subtype. These clinically aggressive SPNs feature diffuse cell sheets with increased nuclear atypia and abundant mitoses, with reported cases containing foci of sarcomatoid (spindle cell) carcinoma. 356
SPPNPs consistently demonstrate nuclear/cytoplasmic expression of β-catenin and often E-cadherin. 357 Additionally, they express cyclin D1, vimentin, PR, CD10, CD99 (dot-like), CD56, claudin-5, claudin-7, and α1-antitrypsin. Cytokeratins are variably expressed, with up to 70% of cases showing positivity depending on antigen retrieval methods. Approximately 50% of SPPNPs express KIT (CD117) but lack KIT mutations. 358 Synaptophysin and neuron-specific enolase may exhibit focal positivity, while SPNs are typically negative for chromogranin A, trypsin, and CEA. 357 Solid SPNs may histologically resemble well-differentiated NENs or acinar cell carcinomas, 99 and the diagnosis in such cases relies on the nuclear β-catenin expression and the absence of labeling for chromogranin A, trypsin, and/or BCL10.
SPPNPs almost universally present alterations in the Wnt pathway. More specifically, mutations in CTNNB1 (which encodes b-catenin) are found in 83–100% of SPPNPs. 100 These are usually missense exon 3 mutations that lead to loss of the glycogen synthase kinase 3-b (GSK3b) binding site, impairing the degradation of the b-catenin and fostering the activation of the Wnt pathway.100–103 This leads to increased expression of multiple members of the Wnt pathway, along with the upregulation of cell cycle regulators, such as CCND1.359,360
Overall, SPPNPs have low TMB and few copy number alterations, 101 and this perhaps explains the rather indolent disease behavior witnessed in most patients. However, a subset of up to 15% of SPPNPs present more aggressive clinical behavior – so-called malignant SPPNP. 102 Limited data suggest additional molecular alterations, such as mutations in cell cycle regulators (TP53 or RB) or loss of epigenetic regulators (BAP1, TET1, or KDM6A), are needed to trigger its full-blown malignant potential.102,361 Additionally, mutations in other genes such as BRCA2, ATM, PIK3CA, PTEN, ESR1, and CDKN2A have been described, some of them successfully addressed with specific targeted therapy – 29% of SPPNPs have potentially actionable mutations.103,173 As expected, malignant SPPNPs display increased levels of copy number alterations when compared to their benign counterpart and dMMR/MSI-H has been anecdotally described in SPPNP.103,362
Surgical resection is the single most important prognostic factor for patients with SPPNP.166,167 Indeed, even patients with more advanced disease, with evidence of vascular invasion (with or without need for vascular resections) and metastatic disease seem to benefit from resection.104,167,168 Also, multiple resections for metastatic relapse have been performed at times, with excellent clinical outcomes, 169 and even liver transplantation in the setting of unresectable metastatic disease has been done with success.363–366
On the other hand, patients with less aggressive diseases might be managed, at least in the beginning, with less aggressive approaches. For patients with asymptomatic small (<2 cm) non-invasive tumors, initial clinical and radiological surveillance might be adequate. 367 However, as patients are generally young, this only postpones surgery (for an average of 12 months). 95 Additionally, for patients with non-invasive disease, tumor enucleation and other limited resections have been shown to lead to good surgical and survival outcomes in multiple studies, especially in Asia.94,96,368,369 Limited resections have been advocated as they spare pancreatic parenchyma, and therefore, preserve endocrine and exocrine functions. Nonetheless, limited data from the West suggest limited resections might lead to increased rates of tumor recurrence, 92 suggesting one should carefully select patients for this surgical approach.
On average, relapse rates for resected SPPNP are low at 3% (ranging from 0% to 20%). 90 Therefore, the potential benefits of adjuvant chemotherapy are very likely offset by its risks and side effects. Indeed, data from population-based studies suggest adjuvant chemotherapy does not improve the clinical outcomes of patients with resected SPPNP. 167 Also, given the systemic pattern of these few relapses,94,370,371 there seems to be no indication for adjuvant radiotherapy, even for patients with R1 resection.170,171 Importantly, relapses from SPPNP can occur many years after the resection of the primary tumor. Thus, an extended period of follow-up (e.g. 10 years) is justified in this setting. 92
In the setting of metastatic or relapsed SPPNP, case reports suggest SPPNPs might be sensitive to chemotherapy. Multiple chemotherapy regimens have been used, but so far, the best responses have been seen in patients treated with platinum-based chemotherapy, especially with cisplatin and oxaliplatin.173,174 In one Italian multicentric study, four patients with SPPNP were treated with gemcitabine-based chemotherapy (including two with gemcitabine plus oxaliplatin), and no responses were observed. 115 Therefore, based on the limited data available so far, cisplatin-based chemotherapy regimens might be the most active regimens against SPPNP. 172 For unresectable SPPNP, radiotherapy might be an option.172,372,373 However, given that unresectable SPPNPs usually present as large masses, treatments with radiotherapy at high doses might be difficult to deliver due to concerns regarding nearby organ toxicity. 374 Given the frequent expression of PR and its female predilection, theoretically, SPPNP might be sensitive to hormonal therapy, and spontaneous regression of SPPNP coinciding with menopause has been reported. 375 Indeed, a few case reports have looked at the activity of tamoxifen in metastatic SPPNP and have described long-term disease stabilization and even radiological partial response.175–177 Targeted therapy has been shown to be active in very selected cases of metastatic SPPNP. More specifically, patients with molecular alterations in the AKT/PI3K (phosphatidylinositol 3-kinase)/mTOR (mammalian target of rapamycin) pathway treated with the mTOR inhibitor everolimus experienced objective response to treatment.173,178,179 Given the high frequency of b-catenin alterations, it is sound to investigate inhibitors of the Wnt pathway in SPPNP. In this regard, a phase I/II trial is currently investigating the role of tegavivint, a drug that selectively disrupts the interaction of β-catenin and TBL1/TBLR1, in patients with multiple malignancies, including SPPNP (NCT03459469).
After surgical resection, there are controversial findings regarding the risk factors for disease recurrence. The WHO Digestive System Tumor Classification suggests the malignant potential of SPPNPs cannot be predicted by angioinvasion, perineural invasion, or deep invasion of the surrounding pancreatic parenchyma. 99 However, many studies ratify the importance of such features in the patient’s chance of disease recurrence (especially angioinvasion).94,97,371,376 Other clinical and pathological factors have recently emerged as important predictors of disease relapse or survival, such as tumor size (>8–10 cm),94,370,371,376 increased Ki-67 expression,370,371,377 stage IV disease,94,166,376 and male sex.166,378,379 Interestingly, only a few studies have established positive surgical margins and positive lymph nodes as risk factors for disease relapse. 379 Specifically in the setting of metastatic disease, patients with peritoneal disease seem to fare poorly. 94
Conclusion
Rare histological subtypes of pancreatic cancer encompass a group of heterogeneous malignancies, some of which share molecular alterations with PDAC. The identification of these entities is important, as they might display different disease courses when compared to conventional PDAC. Additionally, some of these tumors have completely different molecular backgrounds, suggesting novel therapeutic approaches could be explored. Finally, a remaining challenge is to be able to set up cooperative study groups to evaluate innovative therapies for these rare malignancies.