
Abstract
Epigenetic alterations, including aberrant DNA methylation, are now recognized as bone fide hallmarks of cancer, which can contribute to cancer initiation, progression, therapy responses and therapy resistance. Methylation of gene promoters can have a range of impacts on cancer risk, clinical stratification and therapeutic outcomes. We provide several important examples of genes, which can be silenced or activated by promoter methylation and highlight their clinical implications. These include the mismatch DNA repair genes MLH1 and MSH2, homologous recombination DNA repair genes BRCA1 and RAD51C, the TERT oncogene and genes within the P15/P16/RB1/E2F tumour suppressor axis. We also discuss how these methylation changes might occur in the first place – whether in the context of the CpG island methylator phenotype or constitutional DNA methylation. The choice of assay used to measure methylation can have a significant impact on interpretation of methylation states, and some examples where this can influence clinical decision-making are presented. Aberrant DNA methylation patterns in circulating tumour DNA (ctDNA) are also showing great promise in the context of non-invasive cancer detection and monitoring using liquid biopsies; however, caution must be taken in interpreting these results in cases where constitutional methylation may be present. Thus, this review aims to provide researchers and clinicians with a comprehensive summary of this broad, but important subject, illustrating the potentials and pitfalls of assessing aberrant DNA methylation in cancer.
Plain language summary
Genes can be silenced by molecular tags being placed on them. This is a normal process that controls when and where genes are available to be used. In some cases this silencing can be incorrectly applied to genes involved in preventing cancer, causing cancer initiation and progression. This review discusses the role of one of these tagging processes, DNA methylation and its role in initiation of cancer and implications for treatment.
Keywords
Introduction
It is now well accepted that carcinogenesis and cancer proliferation can be driven by both genetic and epigenetic events. Genetic changes directly affect the sequence of the DNA in a cell, whereas epigenetic changes refer to reversible modifications to DNA, histones or RNA and can affect the structure or function of DNA. Thus, epigenetic changes do not affect the sequence of the DNA or RNA, but do influence the way that genetic information is read and translated by cells. Some epigenetic modifications to DNA can be passed to daughter cells, and in some cases across generations in organisms. 1 DNA methylation at the fifth carbon of cytosine (5-methylcytosine; 5mC) is a well-studied and highly conserved epigenetic modification that is essential for development in mammals. It can influence gene expression levels, protect cells from repetitive element activity, maintain genome stability during cell division and enable parental gene imprinting. In mammals, cytosine methylation primarily occurs at CpG sites in the DNA sequence.2,3 Although these CpG sites occur at low frequencies across the mammalian genomes, the majority (60–80%) of them are methylated. 4 Less than 10% of CpG sites are found in CpG dense regions of the genome, known as ‘CpG islands’, which are often found at transcription start sites of genes. In somatic cells, many CpG islands are unmethylated and this state is associated with the potential for gene expression. 5 However, in the context of cancer, this balance shifts leading to global hypomethylation of the genome coupled with acquired methylation of certain CpG islands. 5 This can lead to silencing of tumour suppressor genes, decreased genomic stability and, in some cases, activation of protooncogenes that can drive carcinogenesis.5 –10 Silencing of tumour suppressor genes via promoter hypermethylation are also described as ‘epimutations’. 11 Indeed, these epigenetic changes can have similar effects to genetic mutations in the process of cancer development, progression and therapy responses. Specific patterns of CpG methylation have also been associated with different cancer types, and these have been used as biomarkers for cancer detection, prognosis and therapeutic outcomes.12,13
Herein, we focus on the impacts of tumour suppressor gene silencing by promoter methylation on cancer initiation and therapeutic responses, with examples provided. We also examine the potential use of unique DNA methylation patterns in cancer for disease risk, diagnosis, monitoring and precision medicine.
Aberrant DNA methylation and carcinogenesis
The first report of tumour suppressor gene silencing contributing to cancer formation was in 1989, describing methylation of the retinoblastoma suppressor gene, RB1, in individuals with sporadic unilateral retinoblastoma. 14 Since then, many other tumour suppressor genes have been found to be silenced via promoter methylation in cancers, including MLH1, BRCA1, RAD51C, APC and CDKN2A/p16 (Table 1), and aberrant DNA methylation has been accepted as a feature across multiple cancer types. 15 Like genetic mutations in these genes, epigenetic silencing appears to be enriched in certain cancer types consistent with patterns of genetic mutations, suggesting that both tumour suppressor silencing and mutations can drive carcinogenesis. For example, MLH1 silencing is found primarily in colorectal 16 and endometrial cancers17,18, BRCA1 and RAD51C methylation in ovarian and breast cancers, and APC methylation in gastrointestinal cancers. 19 Hypermethylation of CDKN2A/p16 promoter leads to inactivation of this gene in oesophageal adenocarcinoma. 20 A more comprehensive list of genes and associated cancers is provided in Table 1. Certain proto-oncogenes can also be activated by loss of promoter methylation, for example, BCL2 in B-cell CLL 6 and NFATC1 in B-cell CLL, 7 and this phenomenon is discussed in more detail by others.8 –10 However, silencing of the TERT oncogene is, in contrast, associated with promoter region hypomethylation.21,22
Tumour suppressors known to be silenced by DNA methylation in various cancer types. This non-exhaustive list describes some well-defined tumour suppressor genes silenced (or in the case of THOR, activated) by DNA methylation (either CpG methylation in the promoter or early exons of the gene) across various human cancer types.19 –64

When it comes to tumour suppressor methylation and carcinogenesis, there tends to be a bidirectional interaction between genetic and epigenetic aberrations. Genetic mutations can drive a dysregulated epigenetic landscape, whereas DNA methylation of tumour suppressors can itself be mutagenetic and induce genetic alterations. 66
Methylation of mismatch DNA repair genes
MLH1 and MSH2 are important genes in the mismatch DNA repair (MMR) pathway, and their deficiency leads to a specific pattern of genomic hypermutation called microsatellite instability (MSI). Approximately 30% of endometrial cancers, 65 and 15% of colorectal cancers67,68 exhibit MMR deficiency. Loss of MMR can happen via germline or sporadic mutations in critical MMR genes, and in the case of MLH1 and MSH2 this can also occur via promoter methylation and gene silencing (Figure 1).69 –73 In colorectal cancer (CRC), the frequency of MLH1 promoter methylation is approximately 20% and is particularly associated with right-sided tumours in elderly women. 70 MLH1 and MSH2 methylation have also been identified in patients with the inherited cancer syndrome Lynch syndrome.71 –73 The increased MSI resulting from loss of these genes can drive genomic instability and cancer development. 74 MMR is responsible for repair of mismatched DNA bases, short insertions or deletions that can occur during DNA replication. 69 These types of errors are more likely to occur at repetitive regions of the genome, including microsatellites or short tandem repeats. 69 Once MMR is lost, there is an increase of these errors in microsatellites across the genome, leading to mutagenic MSI, which drives cancer development and progression.69,75

MLH1/MSH2 gene silencing causes MMR deficiency and drives tumour formation. Loss of MMR via promoter hypermethylation and gene silencing or MLH1 or MSH2 causes a form of genomic instability called MSI. This can also be observed for cases with mutations in these genes or other MMR genes PMS2 and MLH6. MSI creates mutations throughout the genome that can drive cancer formation, with colon, gastric and endometrial cancers frequently observed to have MSI. Lynch syndrome due to constitutional epimutations is observed in rare cases. 76 However, MSI can also generate neoantigens that are presented on the tumour cell surface and make these cancers susceptible to immunotherapies.69,77,78
MSI resulting from MLH1 or MSH2 methylation can, however, have some positive impacts on patient outcomes. High MSI has been demonstrated to have prognostic value in early stage (II) colorectal cancer. 79 It has also been shown to sensitize cells to immune therapy by generating neoantigens that are presented on the surface of cancer cells, distinguishing them from normal cells by the immune system (Figure 1).77,78 Thus, while methylation of tumour suppressors can have implications for the development of cancer, it can also harbour positive prognostic and therapeutic value in some cases.
Methylation of DNA repair genes involved in homologous recombination
Contrasting implications of tumour suppressor gene methylation can also be observed for the homologous recombination (HR) DNA repair genes BRCA1 and RAD51C. BRCA1 promoter methylation (meBRCA1) and RAD51C promoter methylation (meRAD51C) are detected in up to 11% and 2% of ovarian cancers, respectively.23,24 MeBRCA1 has also been found in approximately 3% of breast cancers, 27 and in the triple-negative breast cancer (TNBC) subtype this can increase up to 22%. 26 MeRAD51C may also be found in up to 14% of BRCAmutation wildtype TNBC (EMBRACE clinical trial80,81). Thus, in these cancer types, meBRCA1 and meRAD51C represent a significant proportion of patients.
The HR pathway is critical for the high-fidelity repair of DNA double strand breaks, which are highly toxic events for cells. When this pathway is inactivated by either mutations of the core genes (e.g. BRCA1, BRCA2, RAD51C, RAD51D, PALB2) or silencing of BRCA1 or RAD51C, other more mutagenic pathways of DNA repair compensate. 82 Loss of HR is frequently observed in high-grade serous ovarian cancer and TNBC, usually in the absence of functional TP53 – often described as the guardian of the genome. The resulting genomic instability can drive not only cancer initiation but also cancer progression. However, as observed for MMR genes, silencing or mutation of these tumour suppressor genes can drive cancer on one hand but is also a positive biomarker for response to platinum chemotherapy and targeted PARP inhibitor (PARPi) therapy on the other hand,25,28,83,84 and this is covered in more detail later in this review.
In summary, like HR gene mutations, methylation of BRCA1 and RAD51C can not only serve as a driver of cancer but can also be targeted therapeutically, representing a better prognosis for a proportion of these patients if targeted therapy is implemented.
Methylation of P15/P16/RB1/E2F pathway genes
The P15/P16/RB1/E2F pathway is one of the most frequently altered pathways in cancer and plays a crucial role in regulating cell cycle progression.85,86 CDKN2A (encoding tumour suppressor p16) and CDKN2B (encoding tumour suppressor p15) can negatively regulate CDK4 and CDK6, resulting in hypo-phosphorylation of RB1, which leads to cell cycle arrest. In contrast, hyperphosphorylated RB1 releases activated E2F transcription factor from DNA, thereby initiating DNA replication within S-phase of cell cycle. Thus, P15 and P16 function as tumour suppressors in the late G1 phase of cell cycle, preventing progression to S-phase.87,88 Mutations in this pathway are frequent across various cancer types. 89 Given the importance of this pathway in suppressing tumour formation, a high degree of regulation has evolved. Thus, it is unsurprising that the CDKN2A, CDKN2B and RB1 tumour suppressors can all be silenced by promoter methylation. Methylation of RB1 has been reported in retinoblastoma, glioblastoma, breast cancer and bladder cancer. Methylation of CDKN2A has been reported in oesophageal, gastric, colorectal, pancreatic, lung, bladder, ovarian, breast carcinomas and in melanoma (Table 1). Methylation of CDKN2B has been found primarily in leukaemias and lymphomas (Table 1). We focus on a few examples where the prognostic and therapeutic impacts have been assessed.
RB1 hypermethylation has been found in approximately 15% of retinoblastoma tumours and is a key driver of this cancer type. 32 Hypermethylation of CDKN2A has been detected in 22–52% of non-small cell lung cancers (NSCLC) and has been shown by several studies to have a prognostic value in NSCLC. 90 For example, in one study, patients with hypermethylated CDKN2A had significantly shorter survival (median = 21.7 months) than patients without CDKN2A hypermethylation (median = 62.5 months; p = 0.0001, log-rank test). 91 In Acute Myeloid Leukaemia (AML), CDKN2B methylation has been reported in 49–100% of patients, depending on the population analysed, 92 and has also been found to correlate with poor survival. 93
CDKN2A or CDKN2B methylation can cause increased levels of CDK4/6, which in turn drives hypo-phosphorylation of RB1, suggesting that the CDK4/6 inhibitors may be a useful targeted therapy for CDKN2A/CDKN2B-methylated cancers.94,95 As RB1 is downstream of CDK4/6 in this pathway, CDK4/6 inhibitors would not have an effect in cancers with other mechanisms of RB1 inactivation. Demethylating therapies have been recommended as a therapeutic strategy for AML patients with CDKN2B methylation. 92 This approach has demonstrated success in a colon cancer cell line, where the DNMT1 inhibitor decitabine could cause demethylation of CDKN2A, which appeared to trigger senescence of cancer cells. 96 However, it should be noted that DNMT1 inhibitors like decitabine cause global hypomethylation in cells; thus, it would be difficult to assign their efficacy to demethylation of a single gene or locus.
TERT oncogene promoter methylation
Methylation of the TERT promoter region is an interesting exception to the examples provided so far, in that the methylation of this region leads to increased expression the TERT oncogene, rather than gene silencing. 21 Telomerase reverse transcriptase, encoded by the TERT gene, forms a critical part of the telomerase complex which maintains telomeres but is normally silenced in somatic cells. However, TERT gene expression is activated in ~90% of cancers, facilitating replicative immortality. 97 TERT activation in cancer cells can be achieved via mutations in the promoter region, or methylation of upstream CpG sites. The TERT hypermethylated oncological region (THOR) is a small genomic region containing 52 CpG sites immediately upstream of the core TERT gene promoter. Methylation of THOR is associated with gene expression, whereas unmethylated THOR prevents binding of repressive CTCF and is associated with gene silencing.21,22 THOR methylation has been detected in a range of cancer types, including colon, ovarian, breast, lung, brain, prostate, bladder and blood cancers 21 (Table 1), making it an attractive therapeutic target. Targeted de-methylation of THOR using a modified CRISPR-dCas9 system has been demonstrated in breast cancer cells, and this was shown to result in a low-grade phenotype in cell line xenografts. 98 However, the degree of de-methylation using this approach was not optimal. Treatment of cancer cells pre-clinically with the global de-methylating agent 5′-aza-2′-deoxycytidine has also been shown to cause THOR hypomethylation and TERT gene silencing in many cases,22,99 presenting another potential therapeutic avenue for cancers with THOR methylation.
How does aberrant DNA methylation occur in the first place?
Tumour suppressors are not generally silenced in normal tissues, precisely because they are important cellular gatekeepers. So, what goes wrong during human development or cell division that leads to this phenomenon? In some cases it appears that methylation is a critical early step in tumour formation,100 –102 but in other cases oncogenic drivers have been found to alter the epigenome, for example, activating mutations in KRAS in CRC and IDH1 in glioma which drive the CpG island methylator phenotype (CIMP).103,104
It is interesting that certain tumour types are enriched for inactivation of certain tumour suppressors, whether by mutation or promoter methylation. For example, BRCA1 methylation is frequently observed in cancer types where BRCA1 mutations are also frequently observed.25,105,106 This begs the question: is methylation of tumour suppressors a random somatic event occurring during development that simply promotes formation of certain cancers, or a directed event that reflects the silencing programme of the cell of origin.107,108 The latter model is based on the idea that chromatin features of stem and progenitor cells may act as a ‘blueprint’ for methylation patterns that can be adopted by cancer cells, providing them with stem-like features. 107 There appears to be evidence for both models, depending on the gene and cancer context.
The CpG island methylator phenotype
In some cases, tumour suppressor methylation is associated with CIMP, where multiple tumour suppressor genes can be methylated concurrently. CIMP is primarily detected in colorectal and endometrial cancers.109,110 The exact causes of CIMP are not fully understood, 111 but several factors have been described as potential contributors to its development. Certain genetic mutations or alterations have been proposed to lead to the development of CIMP due to their frequent observation in CIMP cancer types. The best example of this is association between BRAF and KRAS oncogene mutations in Colorectal Cancer (CRC) and CIMP.103,112 There is evidence that activating KRAS mutations can drive CIMP via a transcriptional silencing pathway. 103 However, some recent work using ageing organoid models representing a CIMP epigenome showed that in the case of BRAF, CIMP is required for BRAF mutations to form cancer. 113 There is also evidence that IDH mutations can drive CIMP in glioma by modifying histones and increasing global DNA methylation. 104 Thus, it is possible that the order of CIMP versus oncogenic mutations may depend on the gene and disease context, although further studies are required to confirm this.
There are a variety of ways CIMP can develop and drive cancer. Age-related changes in DNA methylation patterns are well known, 100 and CIMP has been associated with older age in certain cancer types. 114 It is believed that cumulative exposure to various environmental factors over time and the gradual accumulation of epigenetic changes might contribute to age-related CIMP. Chronic inflammation has also been implicated in the development of CIMP in some cancers. Inflammatory processes can lead to the recruitment of immune cells and the release of cytokines, which can influence DNA methylation patterns and contribute to CIMP. 115 For example, inflammation caused by Helicobacter Pylori infection can lead to aberrant DNA methylation in gastric epithelial cells which ultimately drives gastric cancer formation. 116 Epstein–Barr virus infection, which is associated with a number of cancer types, including gastric, has also been linked to CIMP;117,118 however, this mechanism may be via overexpression of DNMT1 following infection rather than inflammation.117,119
Although CIMP is always associated with methylation of tumour suppressor genes, tumour suppressor gene methylation is not always associated with CIMP. In fact, in some cases, methylation of a single tumour suppressor gene appears to be sufficient to drive cancer formation. For example, this is observed in cases of constitutional tumour suppressor gene methylation, which is described in the following section.
Constitutional methylation and cancer pre-disposition
Germline mutations in tumour suppressor genes are well-known drivers of carcinogenesis, and there is now a growing appreciation that aberrant methylation of tumour suppressors (i.e. epimutations) in normal tissues can also play a role in cancer formation and pre-disposition.11,102,120 Constitutional methylation describes methylation of specific genes present in normal tissues, confined to one allele and present either in all cells or in mosaic form101,102 (Figure 2). It most likely arises early in development as a somatic event that then expands through different germ layers at varying frequencies and proportions, depending on the time and location of the event 102 (Figure 2). Constitutional epimutations in various genes have been described and linked to formation of various cancer types (reviewed in detail by101,102,105,121). Most epimutations develop independently of changes in the DNA sequence and are referred to as primary epimutations. In contrast, secondary epimutations are a consequence of a genetic change in a cis or trans-acting factor and can thus be heritable if they occur in a germ cell. Secondary epimutations via in-cis genetic changes were initially described in the 1990s for the FMR1 gene, which causes a neurodevelopmental disorder called Fragile X syndrome. Since then, such secondary constitutional epimutations have been described in other genes and for a range of additional disorders, including various cancers.73,121,122 For example, a heterozygous c.-107A>T variant in the 5′ UTR of the BRCA1 gene has been associated with BRCA1 promoter methylation, and found to be dominantly inherited (present in all three germ layers) in two families affected by familial breast and ovarian cancer. 122 In cancer, tumour suppressor epimutations appear to rely on methylation of a single allele followed by inactivation of the wild-type allele, consistent with Knudson’s two-hit model of tumour formation for tumour suppressor mutations123,124 (Figure 2). Pathogenic germline tumour suppressor gene mutations are present soma-wide, and heritable secondary epimutations would be detected in the same way. 121 In contrast, primary epimutations are a somatic event and typically detected as an organism-wide mosaic pattern when arising early in development (Figure 2). Therefore, it is not surprising that constitutional epimutations are associated with increased cancer risk across multiple cancer types and related to a variety of tumour suppressor genes. 105 For example, an association has been observed between constitutional BRCA1 promoter methylation and breast or ovarian cancers with BRCA1 promoter methylation.125,126 In both tumour types, BRCA1 promoter methylation is mainly present in white blood cells of patients with tumours that have the same morphological characteristics as tumours of patients with a BRCA1 mutation (BRCA1-like), implicating methylation as the mutagenic driver.125,126 Indeed, BRCA1 promoter methylation is rare outside of these two tumour types, suggesting that the methylation event indeed influences tumour initiation.105,125 –128
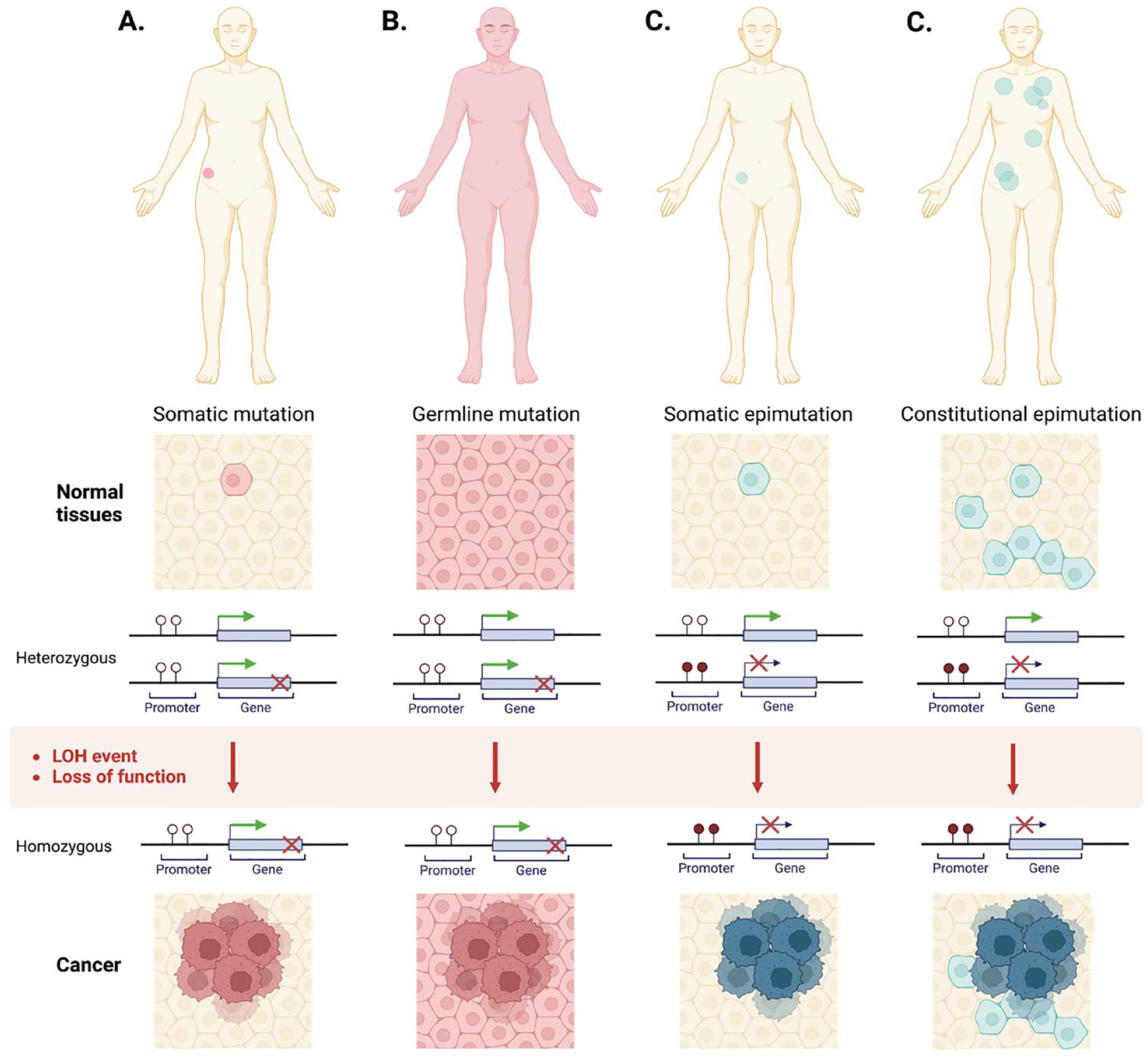
Cancer caused by somatic mutation, germline mutation or constitutional epimutation.
Thus, constitutional methylation of tumour suppressors presents an alternative avenue to cancer formation, independent of CIMP. However, it is still not clear how epimutations arise during development in the first place, and then how these aberrant methylation marks are maintained in cells across generations of cell division. Perhaps the maintenance is a passive mechanism (e.g. DNMT1 simply continuing to replicate a methylation pattern that has formed at random), or some other marks or factors may actively instruct maintenance of this aberrant methylation. 107 It is also possible that both mechanisms are true, but relevant in different tumour suppressor genes or cancer contexts.
Impacts of tumour suppressor methylation on cancer therapy responses
Exploiting silencing of tumour suppressor genes in the clinic
In some cases, silencing of tumour suppressor genes by promoter methylation can have significant impacts on therapeutic responses. One example already discussed is methylation of MLH1 or MSH2 driving MMR deficiency and leading to sensitivity to immune checkpoint blockade. Another example is the impact of BRCA1 and RAD51C methylation on PARPi responses in ovarian and breast cancers.25,28,129
BRCA1 and RAD51C are critical genes in the homologous recombination DNA repair pathway, and mutations in these genes have been shown to sensitize ovarian and breast cancer cells to PARPi. 130 Methylation of these tumour suppressors has been strongly associated with gene silencing, and genomic signatures associated with homologous recombination DNA repair deficiency (HRD), like BRCA1/2 mutated cases.28,81,131 –134 It has been confirmed that meBRCA1 and meRAD51C do indeed predict PARPi responses in ovarian cancer when in a homozygous state, that is, when all cellular gene copies harbour promoter methylation and are completely silenced, leading to HRD.12,25,28 MeBRCA1 has also been associated with good prognosis in TNBC patients. 26 However, homozygous meBRCA1 or meRAD51C can be lost under platinum or PARPi pressure in breast and ovarian cancer, becoming either heterozygous or fully lost.25,28,135 A single unmethylated gene copy of BRCA1 or RAD51C is sufficient to restore HR DNA repair and drive PARPi/platinum resistance in ovarian cancers25,28 (Figure 3), as is observed for heterozygous pathogenic mutations in these genes. Thus, monitoring for real-time quantitative methylation in cancers could help with clinical decision-making.
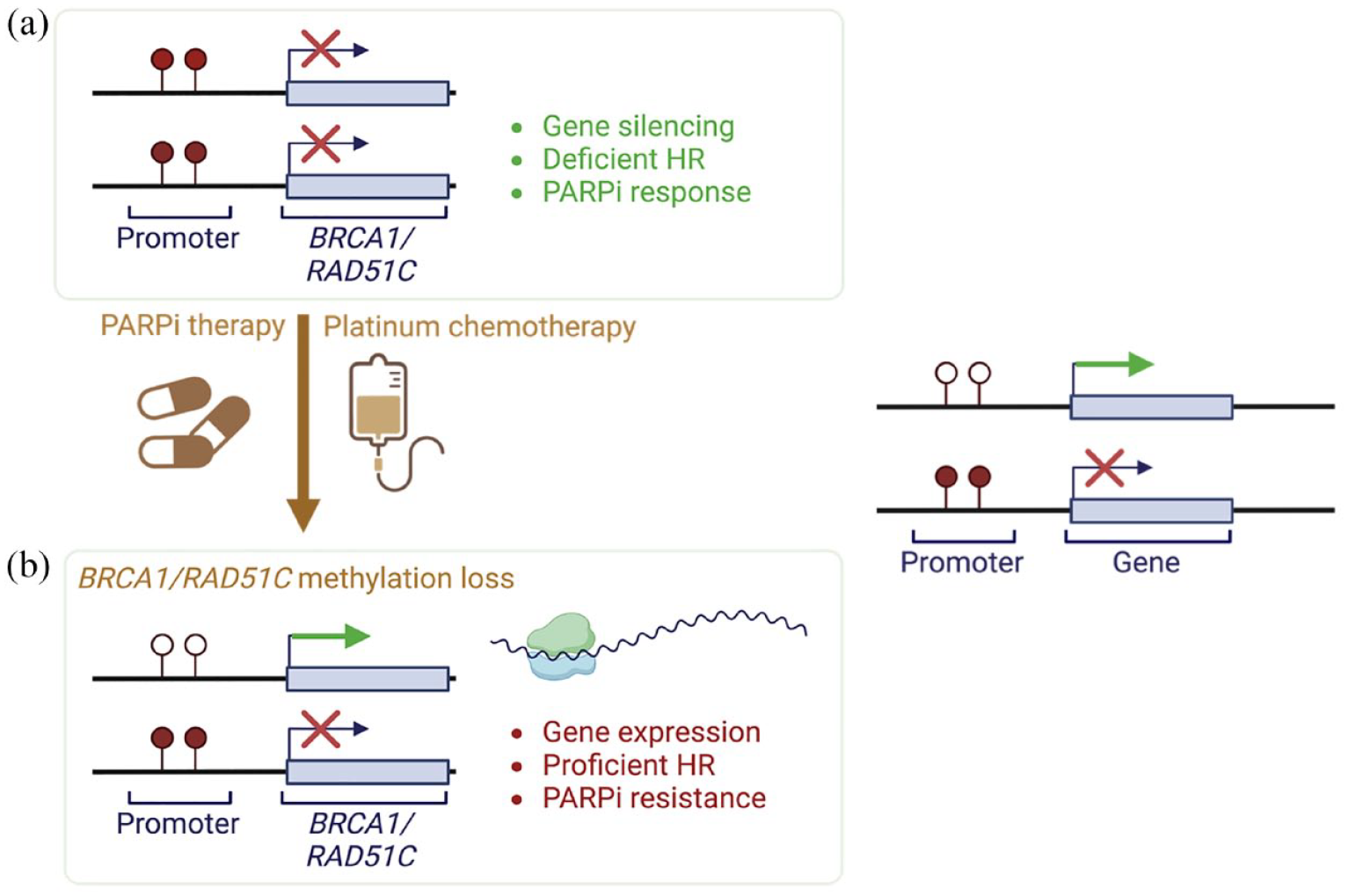
BRCA1 or RAD51C methylation loss and gene re-expression can arise under treatment pressure in ovarian cancer, leading to therapeutic resistance.
An additional example of this phenomenon is methylation of the tumour suppressor and DNA repair protein MGMT (O6-methylguanine–DNA methyltransferase) sensitizing Glioblastoma cells to treatment with alkylating agent temozolomide, 136 with its expression is associated with temozolomide resistance. 137 Thus, the anti-therapeutic effects of de-methylation may occur in additional circumstances.
Demethylation as a therapeutic strategy
In the last decades, various epigenetic modulators have been developed and investigated in preclinical and clinical trials to target DNA hyper- and hypomethylation. 138 Preclinical research has clearly demonstrated the ability of demethylating therapies to decrease tumour suppressor promoter methylation and restored tumour suppressor gene expression, resulting in tumour cell reprogramming and, ultimately, cell death.139 –144 DNA methyltransferase (DNMT) inhibitors (DNMTi) directly impact DNA methylation at a global level within treated cells, whereas other epigenetic therapies, such as histone deacetylase inhibitors (HDACi) and enhancer of zeste homologue inhibitors (EZH2i), can have indirect effects on DNA methylation or gene expression, and are thus often explored in combination with DNMTi in cancers with dysregulated DNA methylation.
As a monotherapy, the most clinically validated epigenetic targeted therapies are DNMTi. 5-azacytidine (5-AZA) and 5-aza-2′deoxycytidine (decitabine). These are cytidine analogues that incorporate into DNA and inhibit DNMTactivity by trapping it, leading to degradation by the proteasome and consequently resulting in hypomethylation during cell replication. 5-AZA and decitabine were approved by the Food and Drug Administration (FDA) in 2004 and 2006 for single agent therapy of myelodysplastic syndromes (MDS) and chronic myelomonocytic leukaemia (CMML).145,146 The relevance of epigenetic treatment in haematological malignancies has been established for years and is reviewed in Santini et al. 147 However, 5-AZA and decitabine have several drawbacks: chemical instability, low bioavailability,148,149 and off-target effects (primarily myelosuppression) due to non-selective inhibition. 150 Thus, their therapeutic use for solid tumours has been limited. To overcome these drawbacks, more stable nucleoside analogues, for example, SGI-110 (guadecitabine), and non-nucleoside compounds have been investigated. Furthermore, a highly potent DNMT1-selective small molecule inhibitor GSK3484862 has recently been developed. 151 In murine embryonic stem cells, this inhibitor caused promoter methylation loss and restored expression of the VIM gene (encoding vimentin), as well as a global decrease in DNA methylation levels with limited toxicity.144,152 However, to date no clinical trials have been undertaken with the new selective non-nucleoside compounds.
The use of epigenetic modulators in multidrug combinational therapies has been shown to improve cancer treatments and potentially overcome drug resistance. Indeed, the synergy of epigenetic drugs and immunotherapies has recently emerged as a very promising field for cancer treatment. 153 For example, results of phase I and II studies have demonstrated that combining nucleoside-based DNMTi with either additional epigenetic drugs, immunotherapies or chemotherapies led to potential benefit in non-small lung cancer154 –156 and ovarian cancer.157 –159 The mechanisms reported to contribute to the synergistic effects of DNA hypomethylating and immune-targeting therapies include modulation of the tumour microenvironment, 160 direct activation of tumour-killing T cells 161 changes in immune checkpoint pathway expression160,162,163 and generation of tumour neoantigens that reveal cancer cells to the immune system.164,165
It should be noted that in the context of meBRCA1/meRAD51C ovarian carcinomas, demethylating therapies, such as DNMT1 inhibitors, might be strategically detrimental for this group of patients, where tumour suppressor methylation is a positive biomarker for PARPi responses. This is less likely to be an issue for MMR pathway loss due to MLH1 or MSH2 silencing, as the immune therapies that exploit these defects rely on the existing DNA damage (MSI) caused by MMR loss, and not an active and continued silencing like that for the HR genes. Thus, the use of these agents in patients should be weighed against the benefits of tumour suppressor methylation that can be targeted by other therapies.
Methods for measuring DNA methylation
Most methods of detecting methylated DNA involve bisulphite conversion – a treatment of DNA that converts unmethylated cytosines in the genome to uracils, but leaves methylated cytosines unaltered. Following PCR, the converted cytosines are read as thymine; thus, methylated and unmethylated bases can be differentiated in a variety of assays (reviewed in detail in166,167 and summarized in Table 2). However, additional new technologies are now emerging that can directly read DNA modifications from the native DNA (e.g. Oxford Nanopore long-read sequencing). 168
Summary of methods available for tumour suppressor methylation testing.

A summary of reported methods that can be used to detect or measure methylation of tumour suppressor promoters.
bisNGS, bisulphite next-generation sequencing; COBRA, combined bisulphite restriction analysis; DHPLC, methylation-specific denaturing high-performance liquid chromatography; HRM, high-resolution melt; MALDI-TOF, MALDI coupled time-of-flight mass spectrometry; MeDIP, methylated DNA immunoprecipitation; MS-DGGE, methylation-specific denaturing gradient gel electrophoresis; MS-MS-HRM, methylation-specific high-resolution melt; MS-PCR, methylation specific PCR; MS-SSCA, methylation-specific single-strand conformation analysis; MS-SNuPE, methylation sensitive-single nucleotide primer extension; MSRE/MRE-Seq or Methyl-seq, methylation sensitive restriction enzyme sequencing; RRBS, reduced-representation bisulphite sequencing; SMART-MSP, sensitive melting analysis after real time-MS-PCR.166 –168
When designing a test for methylation of a genomic region, it is important to consider not only which assay to use to answer a given question, but also which region of the gene/genome is most relevant. For example, if screening for tumour suppressor gene silencing via promoter hypermethylation, it is important that the region of the promoter being analysed is the critical portion that leads to gene silencing.169,170 Furthermore, gene promoter hypermethylation does not necessarily imply that gene expression will be affected. For example, unlike BRCA1 promoter methylation, BRCA2 promoter methylation is not associated with gene silencing in epithelial ovarian cancer.171,172 Thus, analysis of BRCA2 promoter methylation will not yield useful information about DNA repair status of the cancer cells. If analysing methylation that is likely to be present at low levels (e.g. constitutional methylation, circulating tumour methylation in liquid biopsies or low purity tumour samples), it is best to opt for highly sensitive and quantitative assays, such as droplet digital PCR (ddPCR) or targeted bisulphite next-generation sequencing (bisNGS). 173 It is also important to consider whether specific information about individual CpG sites within a particular region is needed. 173 This can become important when assessing heterogeneous CpG methylation of a gene promoter, which is a common occurrence in cancer.28,167 DNA methylation can also exist in a heterozygous state, leading to reduced gene expression, but incomplete gene silencing.25,28 Measuring methylation zygosity can be challenging in patient tumour samples due to variable amounts of contaminating normal cells; however, highly quantitative methylation analysis combined with tumour purity and gene copy number estimates can assist with interpretation of results25,28 (Figure 4).

Definitions of promoter methylation states in tumours. (a) For a tumour suppressor to be fully silenced, the critical promoter CpG sites of all gene copies in a cell or tissue must be methylated. (b) If a single gene copy loses its promoter methylation, the gene product can be expressed and regain function. (c) Methylation zygosity describes combinations of epialleles present at the cellular level. ‘Homozygous methylation’ is when all gene copies in a cell have fully/highly methylated promoters (critical CpG sites are methylated) and the given gene is silenced. This includes aneuploid cases where one allele has been lost due and the remaining hemizygous allele is methylated. (d) ‘Heterozygous methylation’ describes mixtures of fully/highly methylated and unmethylated epialleles coexisting within each cell. In these cells, gene expression is active due to the presence of fully unmethylated epialleles, despite the presence of fully methylated epialleles. (e) Cells with no methylated epialleles have full gene expression. In the case of tumour suppressor genes, this is the normal state of non-cancer cells. (f) Methylation patterns describe epiallele diversity at the tumour/tissue level. ‘Heterogeneous methylation’ describes a highly heterogeneous mixture of epialleles with various CpG methylation patterns present in a tumour sample. (g) ‘Homogeneous methylation’ describes a homogeneous mixture of fully or highly methylated epialleles, which are dominated by one particular CpG methylation pattern in a tissue sample. This can include samples with homozygous methylation within their cells.
In summary, measuring and interpreting promoter methylation in cancer is not always straightforward, and care should be taken when selecting technologies and designing assays or experiments to assess promoter methylation. In some instances, different assays can provide complementary information about methylation of a given region, 174 so it is always important to consider the question being posed, the genomic region being analysed and the purpose of the assay.
Tumour suppressor methylation in liquid biopsies
There is currently a growing interest in the use of cancer DNA methylation biomarkers for the screening, diagnosis and monitoring of cancer using blood samples from patients. Indeed, blood tests appear to be preferred by patients when given a choice of invasive and even other non-invasive tests. 175 Most of these tests are based on detection of circulating tumour DNA (ctDNA), tumour proteins, exosomes or circulating tumour cells and present with multiple advantages over conventional tissue biopsies. 176 Circulating tumour DNA (ctDNA) is released into the bloodstream by tumour cells, or circulating tumour cells, through processes like apoptosis, necrosis and active secretion. It can provide valuable genetic and epigenetic information about the tumour, such as mutations, copy number variations, and DNA methylation changes. These, in turn, can be used for non-invasive cancer screening, diagnosis, prognosis and disease monitoring.177 –179
CtDNA methylation is a relatively stable chemical modification that can be detected in degraded and poor-quality DNA samples, like patient plasma or formalin-fixed samples. Given that certain DNA methylation changes are also cancer-specific, and these changes can occur early in cancer development, there has been growing use of DNA methylation as a cancer biomarker in liquid biopsies. 180 CtDNA methylation in liquid biopsies can be analysed as either a single region (e.g. a single gene promoter),181 –184 as a panel of loci (e.g. multiple gene promoters)185 –189 or using genome-wide epigenetic signatures.181,190,191 Some of these approaches have been FDA approved. For example, Epi proColon [available from: https://www.epiprocolon.com/us/.] is an FDA approved kit for the detection of SEPT9 DNA methylation in serum samples. This test is based on real-time PCR of bisulphite converted ctDNA and has demonstrated an overall sensitivity of 90% and specificity of 88% for detecting CRC at all stages in a retrospective cohort of patients. 192 Although the Epi proColon test relies on a methylation of a single gene, other tests analysing multiple genes or global epigenetic signatures in ctDNA also show great promise. An advantage of these types of tests is that they can potentially detect multiple cancer types in a single test, making them ideal tools for multi-cancer early detection (MCED). The methylation-based PanSeer assay (from Singlera Genomics; https://singleraoncology.com/) interrogates 595 regions at high sequencing depth using their proprietary MethylTitan platform and was shown to detect five common cancer types (stomach, oesophagus, colorectum, lung or liver) in 88% of post-diagnosis patients with a specificity of 96%. PanSeer also detected cancer in 95% of asymptomatic individuals who were later diagnosed, though longitudinal confirmation is needed. 187 The PDACatch assay from Singlera is based on the same technology as the PanSeer and has recently been approved by the FDA for identification of pancreatic ductal adenocarcinoma (PDAC) in individuals at high risk for the disease and outperforms existing PDAC blood markers in terms of sensitivity. 188
The Galleri test (from GRAIL; https://grail.com/galleri-test/), in contrast, analyses > 1 million methylation sites in cell-free DNA fragments to detect changes in global methylation patterns that could indicate cancer presence.190,191 This test was developed based on findings from the Circulating Cell-free Genome Atlas study (CCGA; NCT02889978) showing that whole genome bisulphite sequencing outperformed WGS and targeted sequencing of short variants/indels in terms of cancer detection sensitivity, and that machine-learning classifiers could be used to detect cancer and predict cancer signal origin. 193 The resulting test was found to have a 99.3% specificity for detecting cancer 193 and was also able to predict cancer signal origin with 88% accuracy. 194 The test can detect a methylation pattern common to 50 cancer types, making it ideal for general cancer screening. 190 Validation of prospective patient cohorts is also ongoing in the following studies: PATHFINDER (NCT04241796) STRIVE (NCT03085888) and SUMMIT (NCT03934866), and several successful clinical case reports have been recently reported.195,196 The results reported from these studies so far are extremely exciting, for example interim results of PATHFINDER demonstrated cancer signal in 1.5% (62/4033) of individuals screened, with 40/62 of these having reached diagnostic resolution to date. 197 Sensitive and accurate pan-cancer liquid biopsies would have a massive clinical impact by identifying cancer in patients early (improving survival outcomes) and reducing unnecessary invasive testing in healthy people.
It should be noted, that the presence of ctDNA is different to constitutional methylation of a particular tumour suppressor. Although ctDNA is tumour derived and may represent early or late stage cancer, constitutional methylation is derived from normal cells in the body and may only represent an increased risk of developing a cancer. Constitutional methylation may, thus, be a confounding factor in the detection of methylated ctDNA in liquid biopsies focussed on a single gene/region, potentially causing false positive results in patients. This should be a consideration when designing and interpreting results of such tests.
Conclusion
There is a growing appreciation of the impacts of epigenetic changes in the development and progression of cancer. 15 Indeed, ‘Non-mutational Epigenetic Reprogramming’ has recently been added to the Hallmarks of Cancer originally described by Hanahan and Weinberg. 198 Promoter methylation of various genes can have prognostic implications across many cancer types. Interestingly, tumour suppressor genes that are frequently silenced by methylation in certain cancer types also tend to be frequently inactivated by mutations in the same cancer types. This suggests that, like mutations, epimutations may be selected for during oncogenesis in a tissue and pathway specific manner. Amongst other examples of therapeutic implications, evaluation of methylation of BRCA1/RAD51C and MLH1/MSH2 promoters can predict response to PARP and immune checkpoint inhibitors respectively. In the case of PARPi treatment of ovarian cancers with BRCA1 or RAD51C methylation, however, tumour suppressor gene methylation is a therapeutic target that can be lost under treatment pressure.25,28,129 Thus, monitoring of methylation using appropriately designed assays is critical for providing the best guidance for clinical decision-making. Indeed, we have highlighted the importance of appropriate design and selection of assays for measuring methylation, as each platform has its own advantages and disadvantages. With advances in DNA sequencing technologies, it is now possible to detect low levels of DNA methylation in tissue and blood samples. Identification of methylation in ctDNA from non-invasive liquid biopsies is opening exciting opportunities for early detection of cancer, both for specific cancer types and as a pan-cancer screening tool. Multiple assays are currently being trialled in patients, and preliminary reports have been promising.
There are still many important questions remaining. For example, in cases where tumour suppressor gene methylation is a therapeutic target, are there ways in which this methylation could be stabilized to prevent onset of therapeutic resistance? Also, could we prevent epigenetic silencing of tumour suppressor genes in order to avert cancer initiation? An improved understanding of how methylation develops and is maintained in cancer cells could provide answers to these and other critical questions in the future.