
Abstract
Recently, the possibility of using immune gene signatures (IGSs) has been considered as a novel prognostic tool for numerous cancer types. State-of-the-art methods of genomic, transcriptomic, and protein analysis have allowed the identification of a number of immune signatures correlated to disease outcome. The major adaptive and innate immune components are the T lymphocytes and macrophages, respectively. Herein, we collected essential data on IGSs consisting of subsets of T cells and tumor-associated macrophages and indicating cancer patient outcomes. We discuss factors that can introduce errors in the recognition of immune cell types and explain why the significance of immune signatures can be interpreted with uncertainty. The unidirectional functions of cell types should be entirely addressed in the signatures constructed by the combination of innate and adaptive immune cells. The state of the antitumor immune response is the key basis for IGSs and should be considered in gene signature construction. We also analyzed immune signatures for the prediction of immunotherapy response. Finally, we attempted to explain the present-day limitations in the use of immune signatures as robust criteria for prognosis.
Keywords
Introduction
Tumor cells within identical genetic grounds vary in terms of growth, survival, metastasis, and treatment response. The key mechanism underlying such heterogeneity includes the nature of the interaction of tumor cells with the components of the tumor microenvironment (TME), both locally and systemically. 1 The precursors of stromal cells recruited from the bone marrow as well as infiltrating inflammatory immune cells recruited from the peripheral blood or depo constitute the TME that supports the growth and progression of malignant tumors.2,3 This view of the tumor as a complex multicomponent system is a promising avenue for developing new approaches to anticancer therapy and new biomarkers for disease prognosis based on the TME components.
Over the past 20 years, different gene signature assays have been developed and validated in multiple clinical trials to stratify patients into different risk groups. Widely used genomic tests predict the recurrence risk of breast cancer patients of the ER+HER2- subtype, including MammaPrint, 4 Oncotype DX, 5 and PAM50 Prosigna Assay. 6 The application of these gene expression signatures is recommended by oncological professional communities (e.g., American Society of Clinical Oncology and European Society for Medical Oncology) and helps physicians in clinical practice. However, the activity of the genes detected in these assays is restricted to the tumor cell biology, including proliferation, apoptosis, angiogenesis, invasion, and metastasis. No widely used multi-gene expression assay utilizes immune cell markers. 7
Accumulating evidence indicates a correlation between immune cells and cancer outcome. Two extreme functional states in the TME have been described: an anti-TME (Th1-like inflammatory response that suppresses tumor formation) or pro-tumor immunosuppressive microenvironment (Th2-like immune reactions that promote tumorigenesis).8,9
State-of-the art methods of genomic, transcriptomic, and protein analysis have helped identify signatures associated with the disease outcome. 10 Gene signatures with high sensitivity and specificity can be used to categorize cancer patients into high- and low-risk groups to predict disease outcome and treatment response. Recently, diverse prognostic models have been constructed using patient cohorts based on the groups of genes associated with factors such as metastasis, 11 epithelial–mesenchymal transition (EMT), 12 angiogenesis, 13 and ferroptosis. 14
Immune-related gene signatures can predict outcomes in numerous cancers. Inflammatory infiltration by lymphocytes and macrophages, which is a manifestation of innate and adaptive immunogenesis, may be crucial for the development of additional risk score panels for immunotherapy and other combination therapies.
In this review, we collected data on immune signatures based on lymphocytes and macrophages, which are key regulators of innate and adaptive immune responses in the TME. Herein, we describe recently identified immune signatures and their correlation with the follow-up period in patients with different solid tumors. We also analyzed immune signatures for predicting immunotherapy response. Finally, we present our perspective on the possibility of using immune signatures in clinical practice.
Macrophage-related immune signatures
Macrophage infiltration (macrophage-related genes) is associated with poor prognosis in numerous human cancers, including breast, lung, prostate, gastric, and ovarian cancers, as well as melanoma and glioblastoma. 15 Tumor-associated macrophages (TAMs) are major components of the innate immune system in the TME.15,16 TAMs are highly heterogeneous cells that originate from resident tissue-specific macrophages and newly recruited monocytes. Two main directions of TAM polarization are defined: classically activated pro-inflammatory M1 and alternatively activated pro-tumor, anti-inflammatory M2. 17 However, the diversity of TAMs is more complex, and phenotypes are formed by surrounding TME interactions. 15 Multiple studies have demonstrated the macrophage marker gene signature (MMGS) as a prognostic indicator for patient outcome (Table 1).
Macrophage-related gene signatures.

EMT, epithelial–mesenchymal transition; MFS, metastasis-free survival; OS, overall survival; pCR, pathological complete response; RFS, recurrence-free survival.
Immune signatures based on macrophage-derived genes
MMGS was developed based on single-cell transcriptome data from six primary triple-negative breast cancers (TNBCs). 7 The MMGS model consists of SERPINA1, CD74, STX11, ADAM9, CD24, NFKBIA, and PGK1. The MMGS risk score was an independent prognostic factor for overall survival (OS) in multivariate analysis, which was validated in The Cancer Genome Atlas (TCGA) and GSE96058 dataset cohorts. Patients in the high-risk group had significantly shorter OS than those in the low-risk group in both cohorts. 7 In another study, a signature consisting of five hub macrophage-associated genes (CD79A, CXCL13, IGLL5, LHFPL2, and PLEKHF1) divided patients with TNBC into two groups (Cluster A and Cluster B) based on consistency cluster analysis. 18 Cluster A was responsible for a significantly worse prognosis than that associated with Cluster B. 18 A gene signature comprising 15 genes (TNF-α, IL-1β, IL-6, MMP1, MMP9, TGF-β1, TGF-βRII, EGFR, TP53, WTAPP1, SLC12A5, PSAT1, ESR1, TPD52, and PRKCD) indicated an interaction between TAMs and breast cancer cells. 19 This gene signature correlated with high progression risk and poor survival (overall, metastasis-free, and recurrence-free) in many breast cancer datasets. 19 A 12-immune gene signature (IGS) was constructed in breast cancer patients based on the LCAM, CYP1B1, MYBPC2, LCN2, FAM179A, FAM159A, LIMD2, PIGR, RAC2, IL10, CHI3L1, and CCR8 genes, which are enriched in immune processes such as immune response, leukocyte activation, and cell adhesion. 20 The high-risk immune group had a significantly worse OS. The level of M2 macrophages in the high-risk group was significantly higher than that in the low-risk group. Breast cancer (BC) patients with pathological complete response (pCR) after neoadjuvant chemotherapy have significantly lower immune gene scores than those of patients without pCR. 20
Glioma patients from the Chinese Glioma Genome Atlas and TCGA cohorts, who had high expression of macrophage-related genes, experienced remarkably shorter OS. A TAM-based nine-gene signature (SOCS1, MSR1, TGFB1, CTSC, SOCS3, PTX3, FN1, PSME2, and CXCL9) was constructed, and patients were divided into groups with high and low risks of progression. 21 In another study, MScore included 26 macrophage-related genes that were positively correlated with a lower OS probability in glioma patients. 22
In thyroid cancer, a four-gene risk-scoring model is used to establish a prognostic nomogram that independently predicts OS. The immune signature includes SPP1, CFB, DHRS3, and SLC11A1, which are associated with M1 macrophages. Patients in high-risk group had shorter OS compared to low-risk patients. 23
The M1/M2 two-gene signature (IL-1β, M1 marker, and TGF-β1, M2 marker) based on malignant pleural effusion macrophages of patients with advanced lung cancer demonstrated a significant association with patient outcome. 30 Univariate and multivariate Cox proportional analyses have revealed that the high-risk (M2) population have shorter OS and higher mortality rates than those of the M1 population. 30 The analysis of two gene sets associated with the macrophage phenotype revealed a nine-gene signature (KIF23, BIN1, LAPTM4A, ERAP2, ATP8B2, FAM118A, RGS16, ELMO1, and RAPGEFL1) in pancreatic cancer patients. 24 Based on this signature, low-risk patients have significantly longer OS than those of high-risk patients. 24
Immune gene sets associated with macrophage infiltration
Comparison of tumor samples with high and low macrophage infiltration in gastric cancer (GC) patients has allowed the establishment of a signature comprising seven genes (FGF1, GRP, AVPR1A, APOD, PDGFRL, CXCR4, and CSF1R). 25 This signature predicted OS in five independent TCGA cohorts, wherein high-risk patients exhibited higher levels of M2 macrophage infiltration and lower OS than those of low-risk patients. 25 A total of 10 hub immune-regulated genes (IRGs), including S100A12, DEFB126, KAL1, APOH, CGB5, GRP, GLP2R, LGR6, PTGER3, and cytotoxic T-lymphocyte-associated protein 4 (CTLA4), are also prognostic in GC. 26 High-risk patients have shorter survival times than those of low-risk patients. The prognostic signature significantly correlates with the infiltration of macrophages but not with those of other immune cell. 26 In another study on GC, M2 macrophages were also significantly enriched in high-risk patients, as determined by the six-gene immune-based prognostic signature. 27
To study the immune-regulating role of consensus molecular subtype four in colorectal cancer (CRC), six immune genes (PROK1, THBS1, FGF11, CRP, S100A14, and CCL19) have been identified. 28 The immune prognostic signature allows the classification of patients into high- and low-risk groups in terms of relapse-free survival (RFS) and OS. M2 macrophages are significantly enriched in high-risk patients. 28
For esophageal cancer, immune- and stromal-related genes (C1QA, C1QB, C1QC, CD86, C3AR1, CSF1R, ITGB2, LCP2, SPI1, and TYROBP) comprise a prognostic model. All genes are positively associated with M2 macrophages. 29 In head and neck squamous cell carcinoma patients, the activated stromal gene signature is associated with high expression of M2 macrophages and other immunosuppressive components, for example, WNT/TGF-β signaling, but low expression of B-cell clusters, B cell/plasma cell metagene signatures, and cytolytic activity. 31 In contrast, in patients with an ‘exhausted stroma signature’, a high expression of the pro-inflammatory M1 macrophage signature is observed. Compared with the ‘exhausted immune class’, the ‘active immune class’ is a significant prognostic factor for better OS and disease-free survival. 31
Among analyzed gene sets from the macrophage-based IGSs, the only single overlapping genes were found – CHI3L1 (two IGSs in BC and glioma), TGFB1 (two IGSs in BC and glioma), CSF1R (two IGSs in gastric and esophageal cancers), and members of ADAM and SLC gene families (breast and thyroid cancer, glioma) (Summarized in Table 1, marked in bold). CHI3L1 (YKL-40) belongs to chitinase-like protein family and expressed by macrophages.32,33 Multiple studies demonstrated the role of YKL-40 in tumor angiogenesis, tumor growth, invasion, and progression in several cancers, including BC and glioma.34–36 Targeting of YKL-40 resulted in the suppression of tumor growth as well as angiogenesis in vivo and in vitro. TGFβ1 induces M2-polarization in macrophages, while macrophage-derived TGFβ1 stimulates tumor growth and aggressiveness.37,38 Solute carrier (SLC) transporters regulate inflammatory response and glucose-associated metabolism in macrophages; however, the role of SLC-expressing TAMs in cancer is not fully investigated.39,40 The clear role of macrophage-produced ADAM proteins in tumor is also not yet established.41,42
In summary, macrophage-based signatures are strongly correlated with tumor progression and poor survival. These observations correspond to the pro-tumor activity of M2 TAMs, as demonstrated in numerous clinical cohorts of cancer patients. 15
Lymphocyte-related immune signatures
In tumors, T cells play a key role in the development of an adaptive immune response that is not limited to antitumor effects. The involvement of T-lymphocytes in the stimulation of tumor progression has been established. 8 The number of tumor-infiltrating lymphocytes (TILs) is a widely used morphological parameter based on the definition of lymphocytes in tumors. However, this integral parameter does not provide insight into lymphocyte subpopulations. Nevertheless, in clinical practice, the TIL amount is used for the prognosis of the disease course in patients with CRC, BC, and melanoma.43–45 The prognostic landscape of TIL-T and TIL-B cells has been most comprehensively demonstrated across 30 cancer types. 46 Distinguishing between B and T cells is restricted to the analysis of CD20 and LCK expression. High LCK protein levels are associated with unfavorable prognosis in invasive breast carcinoma, prostate adenocarcinoma, urothelial carcinoma, thyroid carcinoma, mesothelioma, pheochromocytoma, and paraganglioma, whereas high CD20 protein levels are significantly associated with adrenal carcinoma, oral adenocarcinoma, and prostate adenocarcinoma. TIL-T and TIL-B cells are prognostic factors only in prostate adenocarcinoma. 46
To study immune signatures and their prognostic value, researchers frequently attempt to decipher the cellular composition of the TME. This indicates the possible significance of certain lymphocyte subsets in tumor progression. In BC, the immune-related signature (IRS) comprising 15 genes (NUP43, LOC220729, SNTN, TNFRSF18, CYP4F11, ATP6V1H, GRHPR, NDRG2, HES5, POM121L1P, ASAH2, CCR9, ARHGAP39, NUMA1, and FAM9C) was established. 47 A high IRS score is associated with a shorter OS. Infiltration of B lymphocytes and CD8+ T cells in notably higher in low IRS patients. 47 A total of 15 immune-related genes (CD226, KLRD1, KLRC4-KLRK1, IL2, KLRK1, ITK, SPN, SLAMF1, CD1C, FASLG, CD40LG, TBX21, IL7, LAT, and ITGAX) in the TME are associated with favorable prognosis in BC patients, suggesting that IRS corresponds to effector T lymphocyte activity. 48
A four-gene signature (ARNTL2, ECT2, PPIA, and TUBA4A), named IPSLUAD, is associated with unfavorable outcomes in lung cancer. In high-risk patients, defined by IPSLUAD, exhausted T cells, natural Treg и Th1 predominated, whereas Th2, Th17, and follicular helper T (Tfh) cells are diminished. 49 A 10-gene model includes ARAF, HDGF, INHBE, LRSAM1, NR1D2, NR3C1, PLXNA1, PML, SP1, and TANK, which help stratify small-cell lung cancer patients into high- and low-risk groups. Patients at high risk exhibit more CD8+ T cells, helper T cells, mast cells, and Tfh, but fewer Treg cells and CD56bright cell infiltration than those of patients at low risk. 50 These data partly contradict the concept of T-lymphocyte correlation with cancer outcome. 9 In a lung cancer study, the prognostic model TMErisk was constructed based on SERPINE1, CX3CR1, CD200R1, GBP1, IRF1, STAP1, LOX, and OR7E47P. 51 A high TMErisk value was a poor prognostic factor for OS. Despite the low immune infiltration in the high-TMErisk group, an increased accumulation of common lymphoid progenitors, T helper 1 (Th1) and T helper 2 (Th2) cells, was observed. 51 In that study, T helper cells with opposing activities had a unidirectional association with prognosis.
For GC, three immune-related subtypes (Immunity_H, Immunity_M, and Immunity_L), according to the ssGSEA score, have been identified in two independent Gene Expression Omnibus datasets. 52 Survival analysis has confirmed a favorable prognosis for the Immunity_H subtype with predominant infiltration of CD4+ T cells, CD8+ T cells, and γδT cells. The ADAM gene decysin 1 from the Immunity_H subtype is highly associated with favorable prognosis. 52
Whole-transcriptome analysis of pancreatic tumor samples has been used to identify three immune infiltrate scores based on CD3, CD8, and CD68. 53 In one study, to determine whether infiltration of these key cell types predicts outcomes, patients were divided into high or low infiltration groups. A high CD3 score correlated with improved OS, whereas CD8 and macrophage scores were not associated with OS. 53 The results highlighted the lack of correlation between cytotoxic lymphocyte infiltration and disease outcome. In melanoma, CCL5, GBP5, GZMA, GZMH, IRF1, LAG3, NKG7, PRF1, and PSMB10 genes are associated with stronger T-cell and immune responses. Higher expression of the gene signature correlates with CD8+ T-cell infiltration and better prognosis. 54 Five prognostic immune cell infiltration-related module genes, including FPR1, CIITA, KLRC1, TNFRSF6B, and WFIKKN1, have been identified in bladder cancer patients. The gene signature shows a significant positive correlation with several immune cells, including CD8+ T cells. Signatures independently predicted 1-, 3- and 5-year survival. 55
Overall, indicated T-cell-specific gene signatures divide cancer patients into high- and low-risk groups, where high expression of prognostic genes is associated with worse prognosis (Table 2). Investigated gene sets contain only few overlapped genes – IRF1 (in IGSs of lung cancer and melanoma) and class of GBP proteins (GBP1 in IGS of lung cancer and GBP5 in IGS of melanoma) (indicated in Table 2, marked in bold). IRF1 is one of the most important transcriptional factors for T-cell differentiation. 56 In tumor, IRF1 induces the Programmed cell death ligand 1 (PD-L1) expression and tumor escape.57,58 Interferon γ (IFN-γ)-inducible guanylate binding proteins, such as GBP1 and GBP5, are regulators of T-cell activation. Despite the role of GBP proteins in cancer remains contradictory, some studies propose to use GBP expression for predicting immunotherapy response.59–62
Lymphocyte-related gene signatures.
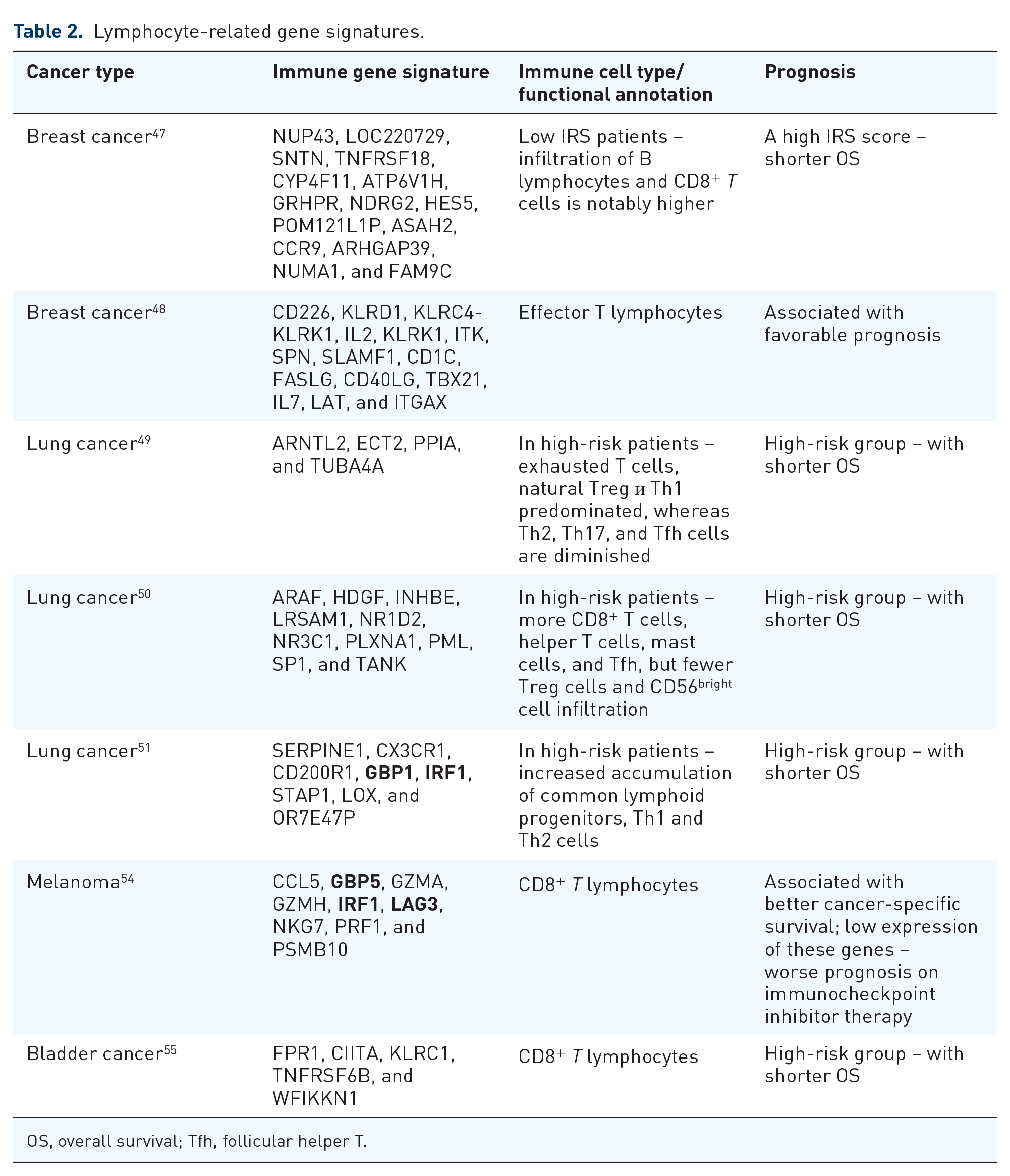
OS, overall survival; Tfh, follicular helper T.
Cell non-specific immune signatures
Most studies on immune signatures, which are not related to specific immune cell populations, were identified for CRC and lung cancer.
In CRC patients, the TME score obtained using the CIBERSORT algorithm significantly correlates with naïve B cells, resting memory CD4+ T cells, and M0 macrophages. 63 The prognosis of CRC patients with high TME scores is worse than that of patients with low TME scores. Three clusters were described according to the TME score: TME cluster 1 (TMEC1) with resting memory CD4+ T cells, TMEC2 with resting NK cells and activated mast cells, and TMEC3 with activated memory CD4+ T cells. 63 In one study on colon cancer, the IRS included genes associated with antigen processing and presentation (LAG3, PSMD11, and TAP2), defense response to infection (CEBPB, CXCL9, IRF8, and RNASE7), epithelial cell migration (ITGB1 and SPARC), and MCFD2. 64 Leukocyte subpopulation analysis indicates that the low-risk signature is enriched with cytotoxic cells (activated CD4/CD8+ T cells and NK cells) and is reduced with myeloid-derived suppressor cells and regulatory T cells (Tregs). 64 The expression of IRGs and the abundance of tumor-infiltrating immune cells (TIICs) in the TME have been compared between KRAS-mutant and KRAS wild-type CRC patients. 65 An immune risk model was established comprising IRGs (VGF, RLN3, and CT45A1) and a TIIC signature including Tregs, M1 macrophages, and activated CD4+ memory T cells. In patients with KRAS mutations, a high abundance of M1 macrophages and activated CD4+ memory T cells is associated with better prognosis, while a high abundance of Tregs is associated with poorer prognosis. 65 The additional immune-related prognostic score consists of 36 unique IRGs. 66 The signature significantly divides the patients into low- and high-risk groups in terms of RFS and OS. 66 M2 macrophage and mast cell infiltration is significantly higher in the high-risk group than in the low-risk group, whereas plasma cell infiltration was significantly enriched in the low-risk group. 66 The two-gene immune-associated RNA-binding protein signature predicts OS and drug sensitivity in colon cancer patients. 67 Plasma cells, CD4 memory resting T cells, monocytes, and dendritic cells (DCs) are downregulated in the high-risk group, whereas M0 macrophages, M2 macrophages, stromal score, and immune score are significantly upregulated in the high-risk group compared to those in the low-risk group. 67
IRGs found in lung adenocarcinoma are associated with immune regulation and biological pathways such as MAPK signaling. The 10-gene immune signature is negatively correlated with B cells, CD4+ T cells, CD8+ T cells, neutrophils, DCs, macrophage immune infiltration, and immune checkpoint molecules Programmed cell death protein 1 (PD-1) and CTLA-4. 68 In non-small cell lung carcinoma patients, the M1 signature, peripheral T-cell signature, and high mRNA expression levels of CD137 and PSMB9 shows better predictive performance than those of known biomarkers, such as PD-L1 expression, tumor mutation burden, or TILs. 69 The M1 signature genes include CBLB, CCR7, CD27, CD48, FOXO1, FYB, HLA-B, HLA-G, IFIH1, IKZF4, LAMP3, NFKBIA, and SAMHD1, whereas the peripheral T-cell signature includes HLA-DOA, GPR18, and STAT1. The progression-free survival (PFS) is significantly longer in patients with high M1 and peripheral T-cell signatures. 69 The 10-IRG pairs signature associated with the tumor immune response was established to predict lung cancer patient prognosis 52. Patients in the high-risk group have poorer prognoses than those of patients in the low-risk group. In the high-risk group, M2 macrophage and neutrophil infiltration are higher, whereas Tfh levels are significantly lower. 70 Based on the five-gene immune score (ANGPTL4, LIFR, SHC3, PLK1, and C6), lung cancer patients in the high-risk group have more CD4+ memory-activated T cells, activated NK cells, M0 macrophages, M1 macrophages, and activated mast cells, whereas CD4+ memory-resting T cells, monocytes, resting DCs, and resting mast cells were more abundant in the low-risk subgroup. Tumor immune dysfunction and exclusion analysis determined that patients in the high-risk group would benefit from treatment with immune checkpoint inhibitors (ICIs) more than those in the low-risk group. 71 In accordance with the above data, not only lymphocytes, but also NK cells and M1 macrophages, demonstrate an association with a poor prognosis in contrast to the basic knowledge on their antitumor activity. 3
GC patients have been stratified into high- and low-risk groups according to 16 prognostic IRGs (HSPA1A, HSPA1B, HSPA5, MICB, PSMC3, TAP2, KIAA0368, RBP1, APOD, VDR, PPP3R1, IL11RA, LGR4, NRP1, PLCG1, and GZMB). The percentages of CD4 memory resting cells and M2 macrophages are significantly higher in the high-risk group than in the low-risk group. In the low-risk group, activated NK cells, memory activated CD4+ T cells, and CD8+ T-cell infiltration are abundant. 72 The eight-IRG signature is an independent prognostic factor for OS in GC patients. In the high-risk group, the fractions of CD4+ T cells, M2 macrophages, and monocytes are higher. The low-risk group has a higher ‘immunophenoscore’, reflecting a better response to ICIs. 73
In hepatocellular carcinoma (HCC), a risk score composed of eight metabolic genes (G6PD, GNPDA1, LDHA, ELOVL1, SLC25A24, CAD, GTDC1, and AMD1) predicted OS in a training cohort (TCGA) and a testing cohort. The high-risk group exhibited obviously higher macrophage accumulation, together with a positive correlation with the expression levels of immune checkpoint molecules (PD1, PDL1, and CTLA4). 74 A five-gene risk score (ATG10, IL18RAP, PRKCD, SLC11A1, and SPP1) was constructed to determine the prognosis of HCC. In the high-risk group, the scores of M1 macrophages, resting mast cells, and CD8+ T cells were significantly lower than those in the low-risk group. 75
An immune signature based on 13-mRNA (SULT1C2, EGFL6, DUSP2, BLNK, BATF, PPFIBP2, HES1, THBS4, CPA3, NEFL, VWF, LRP8, and PDGFD) was established in bladder cancer. 76 Macrophages and Tregs exhibit a strong positive correlation with the immune signature score (ISS), whereas neutrophils show a negative correlation with ISS. Patients with the high-risk subtype have a significantly worse prognosis than that of low-risk patients. 76 Seven identified bladder cancer-specific IRGs (RBP7, PDGFRA, AHNAK, OAS1, RAC3, EDNRA, and SH3BP2) have prognostic significance for OS, cancer-specific survival, and PFS. 77 Increased infiltration of CD4+ T cells, CD8+ T cells, macrophages, neutrophils, and DCs was observed in the high-risk group. In contrast, a higher B cell level was indicative of a low-risk group. 77
Eight immune genes (ITGA7, RBM14, DENND4B, LQK1, ZNF709, COL7A1, SP1, and NCBP2) have been identified as independent prognostic factors for OS in pancreatic cancer patients. The infiltration levels of diverse immune cells, including DC, iDC, pDC, B cells, T cells, Tcm, Tem, Tfh, Th17 cells, and cytotoxic cells, in the low-risk group were significantly higher than those in the high-risk group. 78
A low-risk group of squamous cell carcinoma (SCC) patients, who had high proportions of naïve B cells and resting mast cells, experienced better OS than those of a high-risk group. 61 Genes-immune checkpoint modulators (CD27, TNFRSF14, CD276, IDO1, TIGIT, CTLA4, ICOS, and LAG3) have prognostic value for SCC patients. 79
Five IRGs (BUB1B, NDN, NID1, COL4A6, and FLRT2) constitute the risk signature for prostate adenocarcinoma. The infiltration level of CD4+ memory-activated T cells and Tregs was higher, whereas the infiltration levels of plasma cells, monocytes, activated mast cells, and neutrophils were significantly lower in the high-risk group than in the low-risk group. 80
A four-parameter tumor immune signature based on the infiltration of CD8+ T cells, galectin-9+ DCs, or DC-like macrophages, a high M1/M2 ratio, and a high expression of galectin-3 by the tumor cells formed a positive prognostic factor for long-term survival in patients with stage IV melanoma patients. 81 IRGs (CHIT1, GTSF1L, PLA2G2D, and GNG8) in cervical cancer were positively correlated with OS and immune infiltration, including activated B cells, effector memory CD8+ T cells, eosinophils, and plasmacytoid DCs. 82 In another study, eight immune cells (activated B cells, activated CD8+ T cells, eosinophils, monocytes, activated CD4+ T cells, effector memory CD8+ T cells, immature B cells, and plasmacytoid DCs) were protective factors against OS in cervical cancer patients. 83
Mixed cell-derived immune signatures should provide more comprehensive and stronger prognostic value due to combining the significance of diverse cell types. In most of analyzed IGSs comprised of mixed cell types, high risk correlated with the presence of M2-macrophages, neutrophils, CD4+ T lymphocytes, and Tregs, whereas low risk correlated with CD8+ T cells, B cells, DCs, and NK cells. However, in some IGSs high-risk group included also M1 macrophages, CD8+ lymphocytes, NK cells, and other cytotoxic immune cells (Table 3). It means that the unidirectional functions of used cell types, including innate and adaptive immune cells, should be considered.
Cell-mixed immune gene signatures.
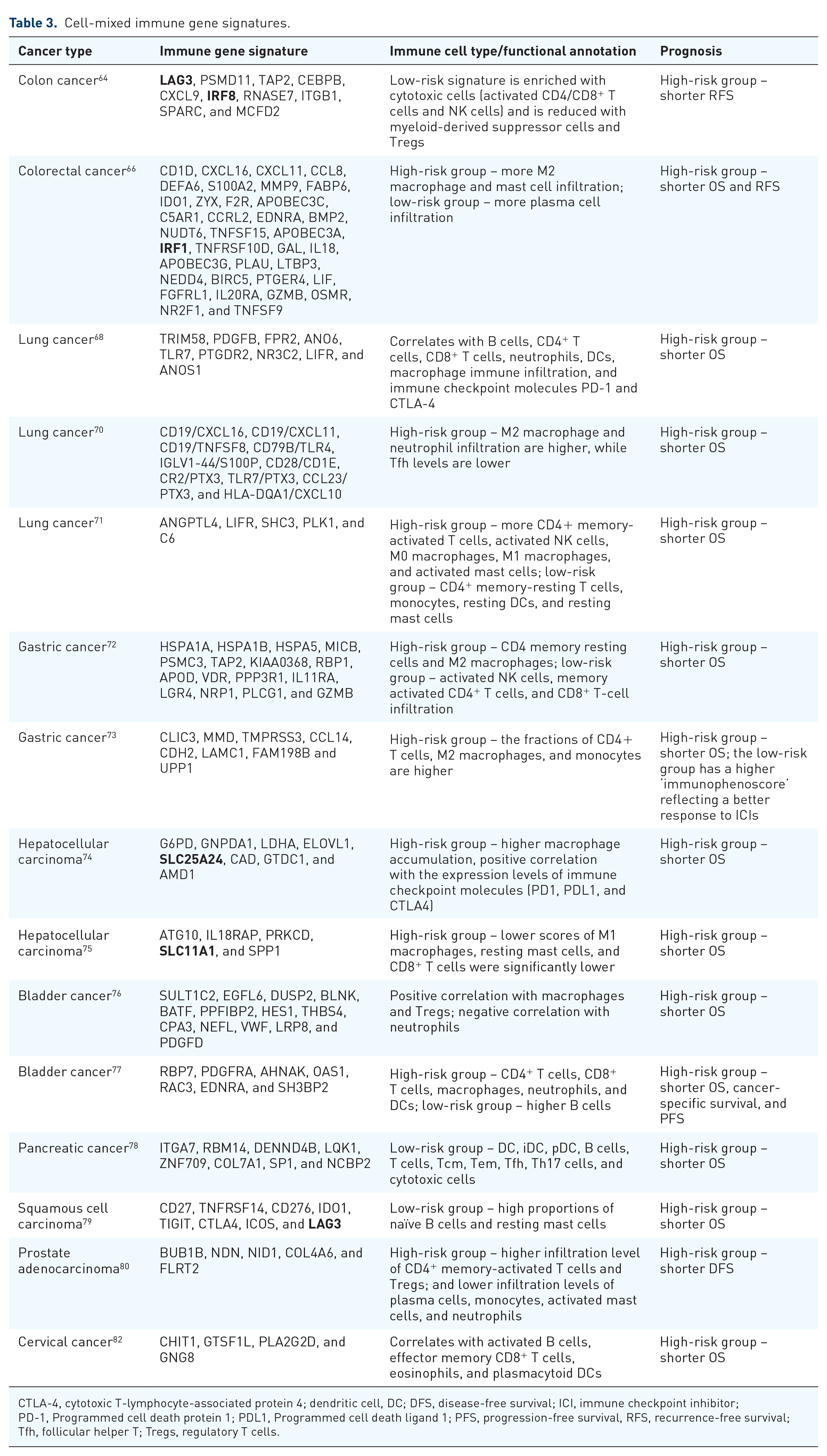
CTLA-4, cytotoxic T-lymphocyte-associated protein 4; dendritic cell, DC; DFS, disease-free survival; ICI, immune checkpoint inhibitor; PD-1, Programmed cell death protein 1; PDL1, Programmed cell death ligand 1; PFS, progression-free survival, RFS, recurrence-free survival; Tfh, follicular helper T; Tregs, regulatory T cells.
Among overlapped genes, we found increased expression of LAG3 in SCC, colon cancer (Table 3) and melanoma (Table 2). LAG3 contributes to T-cell exhaustion and is one of the main FDA-approved ICI targets.84,85 The presence of SLC family genes in mixed cell IGSs correlated with high macrophage infiltration and was also found in macrophage-associated gene signatures (Tables 1 and 3, marked in bold). At the same time, IGSs containing IRF genes positively correlated with T-cell infiltration, whereas T-cell-specific gene sets also include these genes (Tables 2 and 3, marked in bold).
Universal immune signatures not associated with specific cancer type
Extensive immunogenomic analysis of more than 10,000 tumors comprising 33 diverse cancer types was performed using six molecular platforms: mRNA, microRNA, exome sequencing, DNA methylation, copy number, and reverse-phase protein arrays. 86 Across cancer types, authors identified six immune subtypes: ‘Wound Healing’, ‘IFN-γ Dominant’, ‘Inflammatory’, ‘Lymphocyte Depleted’, ‘Immunologically Quiet’, and ‘TGF-β Dominant’, which were characterized by differences in macrophage or lymphocyte signatures, Th1/Th2 ratio, extent of intratumoral heterogeneity, aneuploidy, extent of neoantigen load, overall cell proliferation, expression of immunomodulatory genes, and prognosis. Lymphocyte expression signature A high number of unique T cell receptor (TCR) clonotypes, cytokines of activated Th1 and Th17 cells, and M1 macrophages were strongly associated with improved OS, whereas wound healing, macrophage regulation, and TGF-β were associated with worse OS. C3 immune subtype, defined by elevated expression of Th17 and Th1 genes, correlated with better OS in six tumor types, and C4 with more prominent macrophage signature, suppressed Th1, and a high M2 response correlated with poor OS in three cancer types. 86
A large associative study including 7007 arrays from 32 types of cancers revealed a new T-cell signature, named signature-H cells. 87 The identified signature-H included the following 15 genes: CD2, CD247, CD28, CD3D, CD3G, CD6, GPR171, GZMK, ICOS, ITK, KLRB1, PYHIN1, TIGIT, TRAT1, and TRBC1, overexpressed by T cells. Interestingly, ‘hot’ cancers (lung, stomach, and pancreatic cancers) were highly infiltrated with signature-H cells, but not with signature-B and signature-C cells, which also corresponded to T cells. 87
CD8-based signatures consisting of CCL4, CCL5, CD27, CD276, CD3D, CD8A, CXCR1, CXCL9, CXCL10, HLA-DMB, HLA-DRA, HLA-DRB1, LGALS9, NKG7, TNFSF18, and STING1 were developed for 12 cancer types. The CD8 signature score correlated with the CD8 IHC score across all tumor types, except for pancreatic and prostate cancer. 88
The authors identified a transcriptomic set of 55 differentially expressed genes highly conserved across melanoma, GC, bladder cancer, and renal cell carcinoma (RCC) undergoing T-cell attack (TuTack focused gene set). 89 The 84% of the TuTack-focused gene sets consisted of IFN-γ-related genes. TuTack scores were associated with significantly better OS in melanoma, gastric, and bladder cancer, but no clear correlations were found in RCC. 89 However, the roles of interferons in cancer are contradictory. Type I IFN can protect cancer cells from T-cell-mediated cytotoxicity through the regulation of Serpinb9. 90
Single-cell RNA sequencing (scRNA-seq) of tumors, paracancerous tissues, and blood samples across 21 cancer types was performed. 91 The abundance of exhausted T cells varied dramatically depending on the cancer type. Minimal levels of exhausted CD8+ T cells was noted in basal cell carcinoma and BC, maximal levels – in head and neck, esophagus, and liver carcinomas. For CD8+ T cells, the major tumor-reactive T cells are exhausted T cells. 91 Patients with a higher proportion of terminally exhausted CD8+ T cells had better survival rates than those with a higher proportion of tissue-resident memory CD8+ T cells.
Analysis of 10,062 tumor samples obtained from 32 different cancer types demonstrated that the immune signature based on 382 genes was represented by genes of the Th2 and Th1 pathways. The authors mapped signatures with known clusters and revealed that 20% of the genes refer to T_Cell_cluster, 5% overlap with B_Cell_cluster, and 27% overlap with Th1 cells and macrophages. This signature correlated with a favorable prognosis only in melanoma patients. 92
Interestingly, the results obtained in multiple types of cancer are much stronger criteria that do not contradict the postulates of tumor immunology.
Immune signatures predicted immunotherapy efficacy
Immunotherapy is the most widely implemented therapy in oncological practice, targeting components of TME. 93 Immunotherapy is based on the activity of immune cells in the TME. 94 Immunotherapy is aimed at reactivating immune components suppressed via checkpoints, such as CTLA-4 and PD-L1). 95 Almost 10 years after the introduction of therapeutic options based on ICIs, it became clear that there are limitations to the effectiveness of this class of drugs. 96 This clinical obstacle has encouraged the search for additional predictors to achieve a complete response. The critical point is that the immune system is not sufficient to eliminate tumor cells by itself; otherwise, spontaneous regression would be observed more often in tumors without blockade of the immune response. 97 Therefore, the significance of immune signatures should be considered in the context of immunotherapy.
To date, most studies have focused on CD8+ T-cell infiltration as a predictive biomarker for response to immune checkpoint blockade therapy.98,99 In patients with urothelial carcinoma, the CD8 signature score was an additional significant prognostic parameter for improved PFS and OS, along with PD-L1 expression on tumor cells, serum hemoglobin levels, and presence of liver metastases. 88 The ImmuCellAI study indicated that 24 immune cell types, particularly different T-cell subsets, can serve as predictive biomarkers for a better immunotherapy response. 100
In a cohort of patients with advanced urothelial cancer treated with atezolizumab (an anti-PD-L1 agent), patients with a low-risk ISS exhibited markedly beneficial clinical outcomes and significantly prolonged survival rates compared to high-risk group. 64 The aforementioned immune signature TuTack based on T cells demonstrated that a low score was expected to be associated with elevated tissue immune responsiveness in melanoma and gastric, kidney, and bladder cancers. 89 T-cell-based immune signature-H favorably predicted the response of patients with melanoma to the anti-PD-1 antibody nivolumab. 87 Patients with urothelial carcinoma treated with a PD-L1 inhibitor (atezolizumab), belonging to the high-risk group, had a higher complete response/partial response rate than those in the low-risk group. The risk score was established based on the immune thirteen-mRNA signature (SULT1C2, EGFL6, DUSP2, BLNK, BATF, PPFIBP2, HES1, THBS4, CPA3, NEFL, VWF, LRP8, and PDGFD). 76
A macrophage-related gene signature consisting of 12 genes (ANPEP, DPP4, PRRG1, GPNMB, TMEM26 related to TAMs, and PXDN, CDH6, SCN3A, SEMA6B, CCDC37, FANCA, NETO2 related to tumor cells) was developed based on RNAseq results of TAMs and tumor cells from mouse gliomas treated with BLZ945 (a CSF1R inhibitor). 101 The results were validated in patients with glioma from the CGGA and TCGA cohorts. A total of 12-gene signature predicted the response to anti-CSF1R therapy based on 3- and 5-year survival rates. 101
Conclusions and future perspectives
The prognosis of the disease is important to identify groups of patients that require more frequent monitoring and treatment adjustments. The prognosis is based on the time of survival without metastasis or recurrence or the OS of cancer patients during the follow-up period. This means that prognostic factors can be considered as criteria for predicting resistance to existing postoperative (adjuvant) therapy manifested in the follow-up period.
In this study, we collected multiple examples of IGSs that are indicative of cancer patient outcomes. IGSs are constructed using genes associated with immune system regulation or by a composite of immune cells themselves (Figure 1). Almost all discovered immune-associated gene sets do not have rigorous specificity for immune cell types; therefore, their interpretation becomes a challenge (Figure 1). The use of machine-learning algorithms can introduce errors in the recognition of cell types, and consequently, the significance of immune signatures can be interpreted as uncertain. Moreover, analyzing signatures using not only specific cell types but also the combination of innate and adaptive immune cells, the unidirectional functions of used cell types should be considered. Some of the detected IGSs contradict previously obtained data on the significance of particular cell subpopulations and their known functions in the adaptive and innate immune responses. 8 However, this is a fairly naturally determined trend, as the same cells can have opposite functions in different tumor contexts. This trend was most clearly demonstrated by Sharonov et al., 102 who described the dual role of B-lymphocytes in cancer.
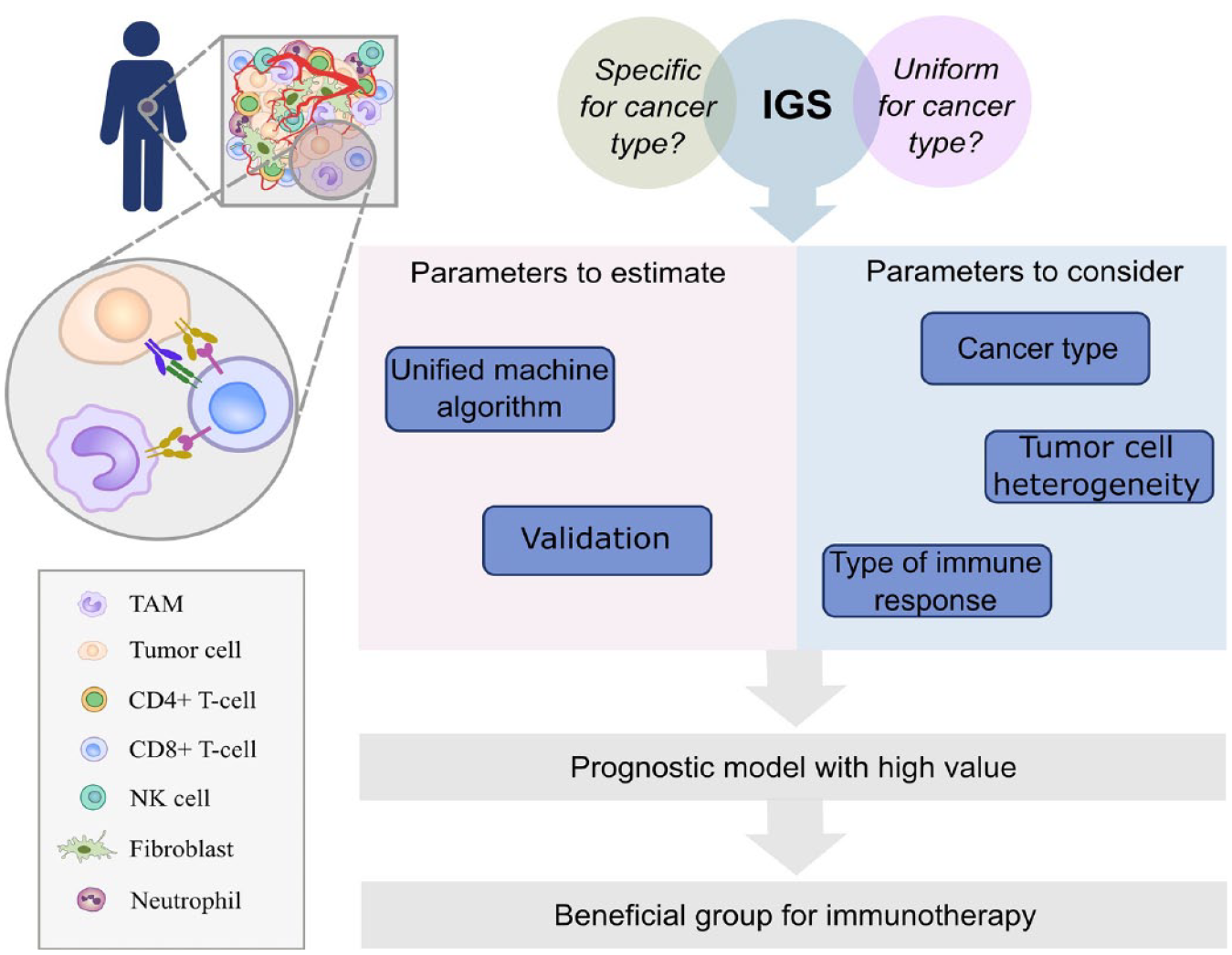
The role of immune signatures in the prognosis of cancer patient outcome. Overall, IGSs can be considered as criteria for predicting the resistance to existing post-operation (adjuvant) therapy manifested in the follow-up period. Should IGSs be uniform for all cancers or specific for cancer type is an open and important question. One of the main limitations of immune signatures is that existing IGSs do not take into account the heterogeneity of the TME, including tumor cell heterogeneity and type of immune response (Th1 or Th2). These parameters together with cancer localization should be considered in IGSs. The use of machine-learning algorithms can introduce errors in the recognition of cell types, and consequently, the significance of immune signatures can be interpreted as uncertain. Unified machine-learning algorithms and appropriate validation should be used. Only after including all these parameters IGSs can get high prognostic value and accuracy. Finally, immune signatures reflecting terminal immune system state in the particular time point can be utilized as criteria to stratify patients for immunotherapy.
Furthermore, described IGSs are not uniform; they do not contain overlapping genes or contain genes with different or even opposite activities (e.g., genes involved in Th1 and Th2 immune responses). They are not properly validated neither in independent cohorts nor in another cancer types. Here, we raise the following question: Should IGSs be uniform or specific for cancer type? (Figure 1).
Tumors are complex and heterogeneous system. 103 However, IGSs do not consider the heterogeneity of the TME along with the heterogeneity of the tumor. By neglecting this fact, we run risks of missing meaningful clinical significance of individual cell populations and, as a consequence, of predicting the course of the disease. Distinguishing more specific subpopulations can help clarify the mechanisms underlying tumor regression or relapse/progression. For example, recently single-cell RNA sequencing revealed a subpopulation of TREM2+ TAMs that were an independent indicator of adverse clinical outcomes in patients with hepatocellular carcinoma. 104
The next question is ‘What is more important – the predominance of particular cell types in the TME or the overall state of the antitumor immune response?’ Unfortunately, this question remains unanswered, and existing immune signatures are not strong enough to address it. Despite the successful application of cancer immunotherapy, clinical experience has shown that even particular patients can respond well to immunotherapy by ICIs, and a large percentage (>50%) do not respond to this therapy. 96 This indicates that the mechanisms of immune escape underlie the incomplete immune response against tumors. The state of the immune system at the onset of treatment and after treatment completion can be a marker of sufficient or insufficient immune response. Spontaneous tumor regression occurs in a state of complete antitumor immune response that has been described in all types of tumors. 105 Microbial infection plays an important role in spontaneous tumor regression accompanied by fever, cytokine release, and immune cell polarization. Th1 cytokines are among the major factors of spontaneous regression of melanoma, 106 while increased amounts of NK cells, as well as CD4- and CD8-positive T cells, are observed in BC. 97
Studying IGSs in terms of their predictive value, efforts should be made primarily to identify possible mechanisms underlying the low potential of the spontaneous antitumor immune response. It is important to understand whether innate and adaptive immune cells in the TME have specificity against tumor cells; otherwise, interventions based on their targeting will not be effective. Restoration of the depleted state of lymphocytes, which is considered the main obstacle for achieving a complete response to immunotherapy, remains unresolved issue. 107
All the aforementioned evidence brings into question the concept that existing immune signatures can serve as robust criteria for prognosis. However, any immune signature can be utilized as a marker of the immune system state at a particular time point during the treatment course. Nevertheless, IGSs associated with a favorable outcome should be considered a distinctive feature of the terminal status of the TME, which must be clinically achieved when immunomodulatory therapy is administered.