
Abstract
Mechanistically, chimeric genes result from DNA rearrangements and include parts of preexisting normal genes combined at the genomic junction site. Some rearranged genes encode pathological proteins with altered molecular functions. Those which can aberrantly promote carcinogenesis are called fusion oncogenes. Their formation is not a rare event in human cancers, and many of them were documented in numerous study reports and in specific databases. They may have various molecular peculiarities like increased stability of an oncogenic part, self-activation of tyrosine kinase receptor moiety, and altered transcriptional regulation activities. Currently, tens of low molecular mass inhibitors are approved in cancers as the drugs targeting receptor tyrosine kinase (RTK) oncogenic fusion proteins, that is, including ALK, ABL, EGFR, FGFR1-3, NTRK1-3, MET, RET, ROS1 moieties. Therein, the presence of the respective RTK fusion in the cancer genome is the diagnostic biomarker for drug prescription. However, identification of such fusion oncogenes is challenging as the breakpoint may arise in multiple sites within the gene, and the exact fusion partner is generally unknown. There is no gold standard method for RTK fusion detection, and many alternative experimental techniques are employed nowadays to solve this issue. Among them, RNA-seq-based methods offer an advantage of unbiased high-throughput analysis of only transcribed RTK fusion genes, and of simultaneous finding both fusion partners in a single RNA-seq read. Here we focus on current knowledge of biology and clinical aspects of RTK fusion genes, related databases, and laboratory detection methods.
Genomic instability and cancer evolution
Genomic instability is an exaggerated accumulation of genomic abnormalities and one of the classical cancer hallmarks.1,2 Genomic instability is associated with both the appearance of ‘driver’ and ‘passenger’ mutations and accelerated molecular evolution of the tumor. There are two main levels of genomic instability. 3 At the nucleotide level, it generates mostly single nucleotide substitutions, short insertions and deletions, insertion of transposable elements, 4 and variability of the microsatellite loci.5,6 In turn, chromosomal instability implies shuffling of the bigger fragments of the genome and is associated with aneuploidy, 7 deletions or amplifications of genes, 8 chromosomal rearrangements, 7 translocations, and gene fusions. 9 Chromosomal instability is currently considered one of the most common features of cancer cells. Approximately 90% of solid tumors and more than a half of hematopoietic cancers have large-scale chromosomal aberrations. 10
Genomic instability generates genomic aberrations and can lead to accelerated evolution of cancers toward drug and immune resistance.11,12 The increased mutation rate can help cancer cells to control drug response by eliminating or amplifying genes related to drug efficacy mechanisms. These processes, underlaying resistance, include loss or gain of chromosomes and their fragments, as well as nucleotide-level mutations. For instance, the reversion mutations of BRCA1/2 in solid cancers make tumors more resistant to platinum or PARPi therapy. 13 Oncogenic mutations can provide immune evasion, such as mutations in KRAS that promote immune suppression in colorectal cancer. 14 Moreover, high level of cancer cells aneuploidy negatively correlates with the effectiveness of immunotherapy. 12 On the other hand, deficient DNA repair systems can also lead to the increased mutation load in protein coding regions, thus expanding the amount of neoantigens in tumor. 15 This makes tumors susceptible to recognition by immune system, thus potentially enhancing immunotherapy efficiency. 16 Thus, high tumor mutation burden is one of the major biomarkers for immunotherapy prescription. 17
Genomic instability is preceded by accumulation of mutations in proto-oncogenes, tumor suppressor genes, or genetic predisposition that lead to defective regulation of tightly connected mechanisms of DNA damage response and repair,18–23 cell cycle progression, senescence, and apoptosis, 24 that coincide with inefficient elimination of transformed cells by the immune system.25–27 For example, negative regulators of Wnt pathway that plays a crucial role in cell proliferation, migration, polarity, and cell fate determination, are usually mutated and, consequently, downregulated in lung cancer cells. 28 This results in abnormal cell proliferation and can provoke metastasis. 28 Immunosuppression can be caused by various random events such as viral infection,29,30 insufficient generation and CD8 T-cell exhaustion, 31 and tissue repair associated with M2 macrophage polarization. 32
Chromosomal instability is associated with an ability to move and interconnect different pieces of chromosomes.33,34 During normal germ cell development, DNA is exchanged or crossed-over between chromosomes during prophase 1 of the meiosis followed by haploid cell division to generate cells with a single copy of each chromosome. These mechanisms are absent in the somatic cells and re-emerge in cancer leading to translocations and aneuploidy.35–38
One of mechanisms that promotes genomic instability is the so-called replication stress, that is, disturbances in the DNA replication process that lead to the arrest or destruction of the replication fork. 39 In turn, termination of replication in the absence of timely repair can provoke DNA double-strand breaks. 40
Hotspots of the DNA double-strand breaks often occur at specific ‘fragile’ sites on chromosomes. 41 At these loci, there is an increased frequency of loss of homozygosity or heterozygosity in cancer cells. 42 In regions of nucleotide tandem repeats, mutations also occur due to malfunctions of the replication fork. 43 The system repairs DNA double-stranded breaks by the homologous recombination of adjacent genome regions. As a result, duplications of microhomologous chromosomal regions can occur, which are found in many types of cancer, such as ovarian and breast cancers. 44
Mitotic disorders are another reason for the appearance of genomic instability at the chromosomal level. Errors can occur both in the early and late stages of cell division. The contribution to the development of genomic instability of sister chromatids pairing violations is well known. 45 Also, mutations in the genes responsible for the passage of cell cycle checkpoints can be associated with genome instability. 46 A common occurrence in the anaphase of cancer cells is chromosome lagging. 47 This is usually due to abnormal attachment of chromosomes, when one kinetochore attaches to microtubules extending from different poles of the spindle. Because this phenomenon is not recognized by the spindle assembly checkpoint, this merotelic attachment and the resulting lagging of the chromosome is an important cause of genomic instability. 46 Mitotic disorders can lead to aneuploidy and even genome-wide duplications. 48 It is important to note that the ploidy of the genome is directly related to the ability of cells to adapt. Tetraploid cells have more mutations per genome, but not per chromosome, which increases the adaptability of the cell. 49 In addition, it has been experimentally shown that genome-wide duplications increase the resistance of cells to subsequent manifestations of genomic instability. 49
Telomere changes can also lead to genomic instability. 50 Thus, shortening of telomeric regions is a frequent event in the early stages of tumor development. 51 Normally, it occurs as a result of the accumulation of the effect of previous acts of progenitor cell replication, and when the threshold value is crossed, such cells undergo aging and/or are eliminated. However, mutations in the cancer cell, for instance, leading to loss of function of negative regulators of cell proliferation, can help to avoid apoptosis. 52 Alternatively, mutations can result in function gain for the different protein families that inhibit apoptosis, for example, members of IAP and Bcl-2 protein families, or other prosurvival factors. 53 This has a strong potential to evade programmed cell death. 54 Telomere shortening can also be associated with various manifestations of genomic instability at the chromosomal level, from amplification or deletion of individual loci to changes in entire chromosomes. 55 Late-stage tumors typically reactivate telomerase or activate an alternative telomere elongation pathway. 50 The latter mechanism appears to be associated with significant genomic rearrangements and defects in DNA repair systems. 56
In addition, high expression and insertions of LINE-1 and other active transposable elements within accessible chromatin regions generate multiple somatic variants, and contribute to the genomic instability in cancers.57–60 Moreover, epigenetic factors can also cause and promote genomic instability. The key epigenetic factors contributing to gene expression regulation and mutagenesis are DNA methylation, histone modification, nucleosome remodeling, and non-coding RNAs. 61
Novel fusion genes in cancer
Chromosomal translocations, deletions, insertions, and inversions can lead to the formation of chimeric genes with oncogenic functions. The first chimeric gene was described within the so-called Philadelphia chromosome, which is formed as a result of the translocation of regions of chromosomes 9 and 22 [t (9;22) (q 34; q 11.2)] and occurs in approximately 90% of all cases of chronic myeloid leukemia.62,63 As a result of structural rearrangement, a chimeric gene BCR-ABL1 is formed, which encodes the corresponding chimeric protein BCR-ABL. 64 It is a continuously active tyrosine kinase that permanently activates proliferation, replication, differentiation, and also renders the cell resistant to apoptosis. 64 Active proliferation can lead to an increased rate of mutagenesis, which contributes to the development of drug resistance in cancer cells. 65
Since the discovery of the Philadelphia chromosome, many other chimeric genes have been found that are specific to a particular type of cancer. 65 For example, the chimeric EWSR1-FLI gene is characteristic for a number of sarcomas.66,67 KIAA1549-BRAF fusion oncogene is characteristic for spinal intramedullary astrocytomas, 68 and fusions with JAZF1 and YWHAE genes frequently occur in leiomyosarcomas.69,70 In many cases, fusion genes are just passenger mutations accompanying carcinogenesis that do not play any significant role in cancer progression.71–73 On the other hand, some fusion genes harbor specific tumor promoting molecular activities and are, therefore, referred to as driver mutations. 74 Their tumor-promoting activities may strongly differ in nature and are still most probably not completely understood.
However, even for the fusion oncogenes, only a tiny fraction of them currently serves as the targets for cancer therapeutics. All such chimeras are fusions with receptor tyrosine kinase (RTK) genes. Presence of such fusion gene in the genome can be a significant biomarker both for the diagnosis and prognosis of the disease, and for the choice of therapy. Tens of small molecular mass therapeutics that inhibit kinase activities of these gene products have been approved by the US FDA for treatment of tumors with confirmed chimeric genes (Table 1). In particular, chimeric transcripts of genes for RTKs ALK, FGFR 1-4, NTRK 1-3, RET, ROS1, and MET are used for prescription of the respective targeted therapeutics (Table 1).
US FDA-approved cancer drugs targeting fusion genes. Only three out of 35 drugs approved for BCR-ABL1 fusion are shown (Data collected from https://nctr-crs.fda.gov/fdalabel/ui/search).

Ph+ CML – chronic myeloid leukemias with Philadelphia chromosome.
NSCLC ALK or ROS1 positive – non-small-cell lung cancers harboring translocations of ALK or ROS1 genes.
ALCL ALK positive – anaplastic large-cell lymphoma, systemic ALK-Positive.
NSCLC ALK positive – non-small-cell lung cancers with translocation of ALK gene.
NSCLC ROS1 positive – non-small-cell lung cancers with translocation of ROS1 gene.
RET-positive NSCLC – non-small-cell lung cancers with translocation of RET gene.
For these fusion oncogenes, all paternal tyrosine kinase receptor proteins have a set of common structural features (Figure 1). They consist of (at least) an extracellular ligand-binding domain, transmembrane domain, and cytoplasmic tyrosine kinase domains.93,94

Life cycle of the tyrosine kinase receptor. Ligand binding to RTK monomer mediates receptor dimerization, autophosphorylation, and various adapter protein binding. In turn, RTK interacting proteins mediate receptor internalization and downstream RAS/MAPK cascade activation including negative and positive feedback loops regulating ubiquitination and phosphorylation. Internalized RTK continues signaling and upon ubiquitination can be recycled to the cell surface.95–98 Alternatively, RTK can be subjected to lysosomal degradation.96,98
Binding of the ligand to the extracellular part of the receptor causes dimerization and transphosphorylation of tyrosine residues of the intracellular domain. This leads to activation of the kinase domain and subsequent triggering of various intracellular cascades. 99 In this case, the signaling pathway of mitogen-activated protein kinase (MAPK pathway), the phosphoinositide-3-kinase/protein kinase B (PI3K/AKT/mTOR), protein kinase C (PKC), and STAT-dependent pathways are activated, which are responsible for the induction of proliferation and cell survival.100–103 Breakage and ligation during chimera formation occur in the intron region and, thus, due to splicing, the exon–intron structure of both parts of the transcript is preserved. Ligand binding leads to autophosphorylation and adaptor protein binding that further phosphorylate and ubiquitinate receptor which is subsequently internalized into lysosomes followed by either recycling or degradation,96,97 Internalized RTK continues to signal inside the cell.
There are two main types of structure of chimeric tyrosine kinase genes (Figure 2). In the first case, the N-terminal extracellular and transmembrane domains-encoding part of a tyrosine kinase receptor gene may be replaced by a partner gene, which results in the presence of tyrosine kinase domain is the 3′-moiety of a chimeric gene. In this case, the 5′-partner domains, as a rule, contribute to the dimerization of the tyrosine kinase domain. For example, the coiled coil motif, sterile alpha domain, LIS 1-homologous, IMD domain, and caspase domain can be mentioned as the contributing structural motifs of the 5′ fusion partner. 104 This can lead to ligand-independent dimerization of the tyrosine kinase domain which triggers further carcinogenic properties of such fusion oncogene.

Structure and functions of the tyrosine kinase gene fusions of the first and second types. 109 (a) Type 1 fusion diagram represents a fusion protein between EML4 and ALK retaining the tyrosine kinase domain, whereas the rest of the RTK including transmembrane domain is lost. The resulting chimera translocates into the cytoplasm where it signals in a RAS/MAPK-dependent manner forming lipid-independent protein granules.110,111 ELM4 is a spindle checkpoint protein 112 whose trimerization domain is retained in chimeras and most likely mediates interaction with the spindle assembly checkpoint complex and mitotic defects.113,114 (b) Type 2 RTK diagram exemplifies FGFR3-TACC3 chimera in which TACC3 dimerization leucine zipper is attached to the C-terminus of FGFR3 mediating ligand-independent dimerization and signaling. In turn, TACC3 is a spindle checkpoint protein and FGFR3-TACC3 chimera causes mitotic defects.115,116
In the second type, the tyrosine kinase receptor is the 5′-terminal partner, and the 3′-partner most probably can additionally stabilize the chimeric RNA or protein product. 105 The breakpoint in such case is usually located after the exons encoding the tyrosine kinase domain. Thus, the partner gene is fused to the C-terminus of a nearly full-length tyrosine kinase receptor. 105
Importantly, for both types, the main feature is the preservation of the active tyrosine kinase domain. This appears to be a key factor distinguishing a ‘driver’ mutation from random chimeric products arisen as a side effect of genomic instability. 106 The second factor is the preservation of an open reading frame for both parts of the chimera. Both factors are important to distinguish between the ‘driver’ and ‘passenger’ gene fusion events. For example, in infant hemispheric glioma, only the patients with preserved open reading frame for a ZCCHC8-ROS1 fusion were responding to entrectinib. 107 Note also the published outstanding case of heavily pretreated glioblastoma which expressed transcripts for four clinically relevant fusion transcripts: with ALK, FGFR2, NTRK2, and NTRK3 genes. Due to tumor heterogeneity, these were, however, expressed each by only a minor fraction of tumor cells, and the prescription of the corresponding targeted therapies would be most likely unsuccessful. 108
Fibroblast growth factor receptor gene fusions
Fibroblast growth factor receptors 1–4 (FGFR1–4) are a highly conserved family of transmembrane tyrosine kinases that can form chimeric oncogenes in various tumors. Normally, FGFR family members play important roles in cell proliferation, embryonal development, organogenesis, maintenance of homeostasis, and tissue integrity. 117 In turn, structural aberrations of FGFR family genes contribute to oncogenesis, tumor progression, and development of drug resistance. In about 8% of the cases, abnormal increase in FGFR activities in cancers is thought to be caused by their gene fusions. 118
Many chimeric gene partners of the FGFR family have been described. 105 The FGFR1-HOOK3 and FGFR1-TACC1 chimeras have been described for cases of the gastrointestinal stromal tumor and gliomas. 119 FGFR1-ZNF703 chimeric gene is found in breast cancer. 120 The products of these chimeras include the N-terminal portion of the FGFR1 protein and the coiled coil motif at the C-terminal, which induces dimerization of the tyrosine kinase triggering various signaling cascades.
Among all family members, FGFR2 gene is more likely than others to form chimeras. 118 As a result of the fusion, the normally prohibited activation of FGFR2 and the launch of various signaling cascades also occur, which can stimulate oncogenesis. FGFR2 fusions are characteristic for cholangiocarcinomas, where they occur in 10–16% of the cases. 121 Thus, in patients with cholangiocarcinoma, the chimeric genes FGFR2–AHCYL, FGFR2–BICC1, FGFR2–PPHLN1, and FGFR2–TACC have been identified and functionally characterized. 104 The chimeric FGFR2-CCDC6 gene can initiate the proliferation of cancer cells in vivo. 122 Furthermore, more than a hundred of FGFR2 chimeric partners have been identified but not studied in detail. Such partners include KIAA1217, KIAA1598, DDX21, LAMC1, NRAP, NOL4, PHC1, RABGAP1L, RASAL2, ROCK1, TFEC, AFF4, CELF2, DCTN2, DNAJC12, DZIP1, FOXP1, INA, KCTD1, LGSN, and other genes. 123 In addition to cholangiocarcinoma, chimeric FGFR2 genes are also frequently found in colorectal cancer, lung cancer, and in hepatocarcinomas.124,125
In turn, FGFR3 fusions are characteristic for glioblastoma, lung, and bladder cancers. 126 One of the best studied chimeras is FGFR3-TACC3. 115 The product of this chimeric gene is a fusion of the N-terminal region of the FGFR3 with the coiled coil domain of TACC3. 115 Coiled coil motif is located at the C-terminus of the protein and is normally involved in the formation and stabilization of the mitotic spindle. 127 The FGFR3-TACC3 fusion has been found in different types of cancer: in gliomas, in cancers of the lung, bladder, head and neck, and cervix.125,128,129 The formation of this chimeric oncogene results in constitutive activation of the FGFR3 tyrosine kinase domain and, as a consequence, activation of the MEK/ERK and STAT1 signaling pathways. 129 Also, the chimeric protein FGFR3-TACC3 is localized to the mitotic spindle and induces errors in the chromosome segregation process, thus resulting in the appearance of aneuploid cells in glioblastoma. 115 In contrast, in bladder cancer cells, FGFR3-TACC3 chimera inhibits TACC3 localization to mitotic spindle, thereby contributing to aneuploidy. 116 FGFR3 fusions with other genes have also been detected but have not been characterized in-depth. For example, the product of the FGFR3–BAIAP2L1 chimera was found in bladder and lung cancers, and the AES, ELAVL3, JAKMIP1, TNIP2, and WHSC1 genes are also known among the confirmed FGFR3 fusion partners.118,130
Fusions with FGFR genes are important prognostic molecular markers. A study was made of tumors of the bile ducts: 152 cholangiocarcinomas and 4 intraductal papillary mucinous tumors were analyzed by fluorescent in situ hybridization (FISH) for the presence of FGFR2 chimeras. 131 Totally, 13 tumors carrying FGFR2 translocations were found which showed statistically significantly longer overall survival: 123 versus 37 months for the patients without translocations. 131 Similar results were obtained in a study of 377 patients with biliary tract cancer, of which 63 had FGFR2 chimeras. 132
Neurotrophic TRK family gene fusions
The neurotrophic TRK (NTRK) family includes TRKA, TRKB, and TRKC proteins which are encoded by the NTRK1, NTRK2, and NTRK3 genes, respectively. 133 Normally, these tyrosine kinases play a pivotal role in neuronal survival and CNS plasticity. 134
Various mutations of the NTRK family genes in cancer cells have been described leading to single nucleotide substitutions, amplifications, and abnormal splice isoforms. 135 However, the formation of chimeric oncogenes is the most common mutation type that leads to increased kinase activity. NTRK gene sequences including a tyrosine kinase domain are usually located at the 3′ end of the chimeras, and are fused to the 5′ region of a partner gene. 136 The product of the chimeric gene is an oncoprotein capable of activating the tyrosine kinase domain without the involvement of a ligand binding. 137 Various NTRK chimeric 5′ partners are known, including ETV6, TPM3, and LMNA, yet their exact roles in the function of an oncoprotein have not been fully characterized. 138
NTRK fusion oncogenes were found in multiple types of cancer. At the same time, their occurrence is relatively low. The analysis of 13,467 adult and pediatric tumor samples from The Cancer Genome Atlas (TCGA) and St. Jude PeCan databases showed that the frequency of chimeric NTRK genes in the most common cancer types was less than 1%. 139 Another study by Rosen and colleagues showed that, when present, functional chimeric NTRK genes are expressed from the very beginning until the most advanced stages of carcinogenesis, thereby indicating a ‘driver’ rather than a ‘passenger’ nature of the mutation. 140
RET gene fusions
RET mutations such as amplifications, single nucleotide substitutions, insertions and deletions, and the formation of chimeric genes have been described in cancer cells. Chimeras with the 3′ RET moiety are more frequently detected and better described. In this case, the tyrosine kinase domain part is usually placed under the control of a stronger promoter, which leads to its overexpression. 106 Many 5′ fusion partners of RET have been described, of which the most common are CCDC6, NCOA4, and KIF5B.103,141,142 RET activation as a result of chimera formation can occur through different mechanisms. First, as already noted, the substitution of the 5′-part of RET for a gene fragment with a stronger promoter. Second, dimerization of RET tyrosine kinase domains due to fusion with partners providing a coiled-coil motif, as in the case of CCDC6 gene. As a result, ligand-independent kinase activation occurs. 143 Also, oncogenic hyperactivation of RET in a fusion is possible due to the loss of the autoinhibitory N-terminal region. 130
ROS1, ALK, and MET gene fusions
ROS1 fusions
ROS1 is a proto-oncogene encoding a tyrosine kinase with unknown physiological function. The first ROS1 chimeric gene was found in the U118MG glioblastoma cell line in 1987. 144 Interestingly, the tyrosine kinase domain of ROS1 has high structural identity with another RTK, ALK. 78 Impaired expression of ROS1 is known for many cancer types, and at least 55 5′-terminal partners of the ROS1 chimeras were reported. 145 The frequency of occurrence of 5′-partners in the chimeras may depend on the cancer type. In particular, large heterogeneity of the 5′-partners is characteristic of glioblastoma, non-small-cell lung cancer (NSCLC), and of inflammatory myofibroblastic tumors. In NSCLC tumors, the most common 5′-partners for the ROS1 chimeras are CD74 (~44%), EZR (16%), SDC4 (14%), and SLC34A2 (10%). 145
ROS1 chimeras are formed due to both intra- and interchromosomal rearrangements. For example, in glioblastoma, the chimeras are more often the result of intrachromosomal translocations, and in NSCLC, they are instead interchromosomal.146,147
The most common structure of the ROS1 chimera includes the loss of virtually the entire extracellular domain of ROS1 and the fusion with the 5′ part of the partner gene, retaining the open reading frame and a complete intracellular kinase domain. 145 As before, the formation of chimeras usually results in ligand-independent, constitutive activation of the ROS1 tyrosine kinase domain. The intracellular localization of the chimeric ROS1 gene depends on the 5′-partner gene and influences the activation of specific signaling pathways. For example, the SDC4-ROS1 and SLC34A2-ROS1 chimeras are localized in endosomes and activate the MAPK pathway more efficiently than the CD74-ROS chimera localized in the endoplasmic reticulum. 148 It was shown in mice models that the presence of the chimeric ROS1 gene alone may be sufficient to induce carcinogenesis. However, when combined with the loss of tumor suppressor p16Ink4a, this results in development of a more aggressive tumor. 149
ALK fusions
Also, many tumors including large cell lymphoma, diffuse large B-cell lymphoma, glioma, NSCLC, colorectal cancer, breast, ovarian, and esophageal cancer have chimeras with tyrosine kinase ALK. 150 More than 90 5′-partner genes of ALK have been described,150–152. ALK translocations are not uncommon and are found in approximately 8% of all NSCLCs. 153 As a result of structural rearrangements, the chimeras retain a full-fledged ALK tyrosine kinase domain at the C-terminus. As in other cases, dimerization of chimeras results in aberrant persistent kinase domain activity. 154 ELM4-ALK is the most frequent ALK fusion in NSCLC. 155 ELM4 is a spindle checkpoint protein required for the proper chromosome alignment and attachment of microtubules to the kinetochore. 112 Accordingly, ELM4-ALK fusion inhibits spindle assembly checkpoint control, that is, inhibits cell cycle arrest in response to the paclitaxel and leads to the mitotic errors (Figure 2), 113,114 This effect is partially due to the kinase activity of ALK domain and, most likely, other oncogenic ALK fusions have acquired kinase activity as well. 114
MET fusions
The MET gene is another important RTK-encoding proto-oncogene. The formation of chimeras with MET is a relatively rare event, but such products are found in various cancer types. The first described chimeric gene was TPR-MET. 156 The replacement of MET extracellular and juxtamembrane domains containing regulatory regions by TPR two leucine zipper domains leads to a constitutively dimerized and therefore activated Met kinase domain. 156 The dimerization domains are essential for TPR-MET oncogene transforming activity, as well as the absence of MET extracellular and juxtamembrane domains. 156 Similar to many other RTKs, the increased MET activity promotes activation of downstream intracellular pathways and signaling axes RAS-MAPK and PI3K-AKT. 157
Structural rearrangements involving MET were detected in 0.5% of cases of NSCLC, 3% of glioblastomas, and isolated cases were described in secretory carcinoma of the salivary gland and pediatric fibrosarcoma.158–161 Several partners of MET chimeras have been described: the HLA-DRB1, KIF5B, PTPRZ1, STARD3NL, and ST7 genes. 162 The stability and degradation of the MET receptor is regulated by an intracellular peri-membrane domain encoded by exon 14. 163 Interestingly, the formation of TPR-MEt also resulted in the loss of exon 14 of MET without disturbing the reading frame. Mutations resulting in the loss of exon 14 in the MET mRNA occur in 3–5% of NSCLCs. 164 This can be caused by both structural rearrangements and point mutations leading to splicing disorders. 165 Mutations are detected both in the intron region around exon 14 and directly in the splicing sites. After pre-mRNA maturation, a mutant MET receptor with an increased lifetime is then translated. 163 These mutations are characteristic of lung adenocarcinomas; however, they are also found in other types of cancer: pulmonary sarcomatoid (pleomorphic) carcinomas, squamous cell NSCLC, and less often in gliomas and other tumors.165,166
Application of protein fusion inhibitors in clinical practice
The recent findings revealed overall better response rate to the approved TKIs in patients with different target RTK fusion genes. 167 Thus, development of new and broader testing of already approved drugs in more than 100 clinical trials targeting 26 gene fusions is currently underway (Table 2).
Drugs tested against neoplasms with gene fusions that are currently in clinical trials (clinicaltrials.org). Please refer to the website for complete information regarding treatment modalities and specific groups of patients in a specific clinical trial.
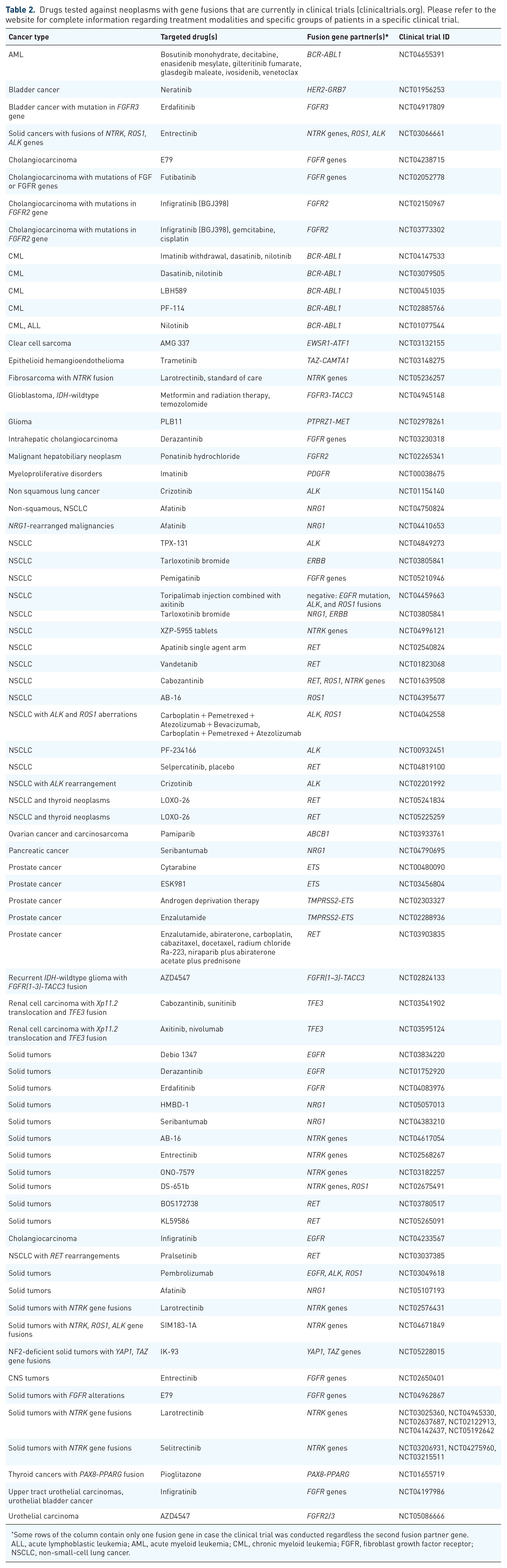
Some rows of the column contain only one fusion gene in case the clinical trial was conducted regardless the second fusion partner gene.
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CML, chronic myeloid leukemia; FGFR, fibroblast growth factor receptor; NSCLC, non-small-cell lung cancer.
Accordingly, the presence of chimeric genes for RTKs is one of the key biomarkers facilitating the choice of therapy. For example, the drug larotrectinib is approved by the US FDA for tumors harboring chimeric NTRK genes. 168 Enterectinib is another drug with a similar profile of action, with somewhat broader activity 85 : it can also inhibit ROS1 and ALK chimeric products. 130 In addition, broad-spectrum tyrosine kinase inhibitors were the first drugs to treat tumors with FGFR chimeras: dovitinib, lenvatinib, lucitanib, nintedanib, derazantinib, and ponatinib. In addition to their activity against FGFR, they can also inhibit VEGFR, RET, KIT, and PDGFR.
However, drugs with low specificity may cause more serious side effects. 169 Therefore, more precise inhibitors have been developed that act specifically against mutations of FGFR family members, including fusion formations. Two selective inhibitors for the treatment of tumors with FGFR chimeric genes have been approved by the US FDA. Erdafitinib is approved for the treatment of urothelial carcinomas with FGFR2 and FGFR3 mutations, which included the chimeric gene FGFR3-TACC3. 170
Selective RET inhibitors pralsetinib (BLU-667, NCT03037385) and selpercatinib (LOXO-292, NCT03157128) in early-stage clinical trials of NSCLC with RET fusions resulted in 56% overall response rate for pralsetinib.88,89 LOXO-292 was recently approved by the US FDA for the treatment of lung and thyroid cancers with RET driver mutations, with respect to fusions. 171 In addition, cases have been described of the acquisition of drug resistance to tyrosine kinase inhibitors in tumors with EGFR mutations, which occurs as a result of structural rearrangements of RET. 172 This also highlights the importance of RET analysis for selecting the optimal treatment strategy.
For the treatment of NSCLC with ROS1 rearrangements, crizotinib has been approved in multiple countries, with an overall response rate of 65–80%. 78 Crizotinib is also the first targeted drug for the treatment of tumors with ALK rearrangements. 79
Entrectinib is an US FDA-approved drug for the treatment of NSCLC with ROS1 rearrangements. 84 Entrectinib is able to penetrate the blood–brain barrier, which is especially important in the treatment of tumors that have metastasized to the brain. Currently, clinical trials investigate the activity of entrectinib against tumors with ROS1 fusions regardless of the cancer type. It was possible to observe objective responses, for example, even for the cases of melanoma and high-grade gliomas.173,174
Crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib are approved by the US FDA for the treatment of ALK chimeric lung cancer. Response to ALK-inhibiting therapy has also been described in studies on renal cell and colon cancers. 173 Of note, a retrospective clinical study showed that progression-free survival after treatment of ALK-positive NSCLC depends on the 5′-partner of the chimera with the 3′-part of ALK. 175 However, the partner genes of the ALK chimeras are not currently routinely taken into consideration when prescribing therapy.
In addition, the drugs capmatinib and tepotinib have recently been approved for the treatment of NSCLC with MET mutations resulting in exon 14 skipping. 175
Experimental methods for identification of fusion oncogenes
Identification of fusion oncogenes is a non-trivial task because there are many alternative methods available that can produce results which may contradict each other (Table 3). It is important, therefore, to carefully select method(s) of choice for the experimental testing considering availability of biomaterials, costs, equipment, high throughput, time of analysis, and even complexity of bioinformatic data interpretation.
Summary of advantages and disadvantages of common methods for detection of gene fusions.

FFPE, formalin-fixed paraffin-embedded; NGS, next-generation sequencing; ORF, open reading frame.
Fluorescent in situ hybridization
FISH is a method based on the hybridization of labeled specific DNA probes. It allows detecting changes in the number of chromosomes, gene fusions, loss of a chromosomal locus or an entire chromosome in both fresh and fixed tumor tissue samples. 176 For example, FISH is used to detect amplifications of the ERBB2 gene for Her2 protein, an important prognostic marker for breast cancer. 177 FISH is also a standard method for detecting the Philadelphia chromosome in chronic myeloid leukemia. 178 In particular, probes for loci that are contiguous in the absence of rearrangements are used to detect translocations. The signal from the native chromosome will be a pair of closely spaced color signals, whereas in the case of rearrangement, the two colors are separated in space. 179
The chimeric EML4-ALK gene is a characteristic biomarker of lung adenocarcinoma. Routine detection of this well-known translocation can also be performed using the FISH method. 180 Likewise, FISH is used to detect known rearrangements of the ETV6 gene in secretory carcinoma and pediatric fibrosarcoma specimens. 181 A number of laboratories use this approach to detect NTRK gene rearrangements and other fusions such as the FGFR3-TACC3 chimeric oncogene.182,183 Nevertheless, FISH diagnostics does not provide information about the functional activity of translocations including preservation of an open reading frame and expression level and does not allow detection of the non-canonical or unknown rearrangements. In addition, this method is poorly sensitive to the detection of local intrachromosomal rearrangements that are characteristic of FGFR2 fusion genes. 184
Immunohistochemistry
Immunohistochemistry ((IHC) method is simple, does not require complex equipment, is relatively affordable, and fast in execution. In addition, reliable results can be obtained using a relatively small amount of starting material. The use of IHC has been approved by the US FDA for the detection of ALK rearrangements in lung cancer samples for selection of patients for treatment with ALK-targeted therapy. 185 Also, a ROS1-specific monoclonal antibody is used to detect ROS1 rearrangements by the IHC. 186
Apparently, IHC method detects not the chimeric gene products themselves, but rather the expression level of the respective tyrosine kinase domain. Thus, increased IHC signal can be registered also due to stronger expression of the whole, non-fusion RTK gene which still can be targeted by the respective specific drug(s). Although this method is easy to perform and inexpensive, its accuracy may be reduced due to technical issues arising from tissue fixation, or other pre-analytical variables. 187 Specifically, comparisons of IHC with FISH and next-generation sequencing (NGS) methods for screening of ALK rearrangements in lung cancer resulted in prevalence of ALK-positive samples tested by IHC.188–190 Furthermore, most of IHC-positive and FISH-negative samples were shown to be negative by NGS, thus supporting the hypothesis of frequent misleading detection of mutations other than fusions (e.g. amplifications) using IHC. 189 However, some patients who were FISH negative but IHC positive for ALK fusion showed complete or partial responses to ALK-targeted therapy, thus supporting the relevance of IHC application in practice 190 and clinical importance of other factors, such as the overexpression of ALK. 191
PCR-based methods
PCR-based methods are widely used both for the diagnosis and for the prognosis and treatment of cancer. Also, reverse transcription PCR is a standard method for confirming the presence of chimeric transcripts resulting from structural rearrangements of chromosomes.178,180 Several kits are commercially available to detect NTRK, ALK, Ret, and ROS fusions in formalin-fixed paraffin-embedded (FFPE) RNA by the multiples real-time PCR using mutation-specific primers and/or hybridization probes with about 1% or more sensitivity and 100% specificity. Detection of 22 fusion genes in AML patients RNA using a commercial quantitative PCR kit demonstrated 99% concordance with cytogenetic analysis. 192 Nonetheless, an agarose gel electrophoresis was used for measurements of multiple EWSR1-ETS and FUS-ETS fusions in Ewing sarcoma by multiplex RT-PCR (Ueno-Yokohata et al. 2021). In turn, high-resolution capillary electrophoresis was used to detect 9 fusion transcripts in the single multiplex reaction. 193 Intriguingly, out of 122 patients examined, abnormal size was detected for one sample due to a rear deletion of the ETV6 exon 5 within ETV6-RUNX1 chimeric gene. Interestingly, RT-PCR was used in Idylia GeneFusion assay to detect fusion products of the NTRK gene without knowledge of the fusion partner by measuring an imbalance between 5′ and 3′ end of the gene. 194 The Idylia GeneFusion assay is basically a device that automatically extracts RNA and performs expression analysis directly from the samples including fresh and FFPE samples. The data demonstrated good concordance between Idylia GeneFusion, IHC, and RNA-seq based Oncomine Focus Assay. Another method, droplet digital multiplied PCR, measures the presence of RNA in small droplets, and each of them encompasses a single RNA molecule, allowing ‘YES-NO’ quantification of PCR results. Thereby, RNA is measured by counting positive and negative droplets. This method was recently used to measure gene fusions from as little as 1 pg of RNA with great success.195,196
To conclude, traditional and novel PCR-based tools represent a gold standard for fusion detection in cancer and are widely used in clinical practice, research, and development.
NGS methods
NGS is an increasingly common method for analyzing tumors. It has a number of advantages over the approaches listed above. Thus, NGS is a more accurate and sensitive method that allows one to analyze various genetic variants in one experimental procedure, eliminating the need to conduct multiple tests in sequence to identify significant biomarkers, thereby significantly reducing the time for selecting therapy.5,197–199 It should be noted that in order to obtain informative results, NGS technologies require a minimum amount of tumor material; they are also able to capture DNA variants present in a minority of cells. 200 To date, many approaches have been proposed for the detection of molecular markers of tumors based on NGS. Although whole genome studies are not widely used in clinical practice, in particular due to their high cost, whole exome analysis, gene panel analysis, and whole transcriptome technologies are increasingly used in the selection of cancer therapies. Methods based on NGS have been developed to search for single nucleotide substitutions, deletions, and duplications of genes, to assess the mutation load of tumors, structural rearrangements of chromosomes, as well as to analyze the level of expression of clinically significant genes and to detect chimeric transcripts.201,202
Several clinical trials investigating applicability of NGS for prognosis and ultimately for treatment decision can be mentioned including Strata PATH (NCT05097599), GITIC Study (NCT02013089), The MATCH Screening Trial (NCT02465060), MyTACTIC (NCT04632992), and BALLETT (NCT05058937). The methodology for detection of mutations used in these studies is based on the NGS. The aim of these studies was formulated as to determine the presence of molecular targets for the approved drugs (including gene fusions) and to verify whether patients will benefit more from the targeted treatment.
DNA sequencing
Whole genome sequencing (WGS) is the most comprehensive platform for genome profiling. 203 The method has potential to identify the genomic locations of all known and currently unknown fusion events. 204 However, application of WGS in tumor biology is nowadays limited to research field due to high costs, lack of standardization, and median turnaround times.
Methods based on whole exome sequencing (WES) are used to detect deletions and duplications of genes, assess the mutational load of tumors, and determine the MSI status. 205 Whole exome studies mainly cover only coding regions of the genome (less than 2%), which provides a reduced cost compared to whole genome analysis with sufficient coverage. Despite technical difficulties, the method is widely used to determine various changes in the genome. For example, several approaches have been developed to detect changes in the copy numbers of specific genome regions [copy number aberration (CNA)] using sequencing data. 206 Perhaps the optimal approach for the analysis of WES data for CNA is measuring of the sequencing depth of different genome regions. 206 If, after normalization, an imbalance in the number of reads in certain part(s) of the genome is detected, then this can be interpreted as a marker of a copy number alteration in the corresponding chromosomal region(s). 206 However, this algorithm cannot detect fusion genes. Moreover, WES as a basis for fusion detection tool has a strong technical limitation, namely its focus on gene exons, whereas most of fusion junctions are located in introns. Thus, WES capacity to detect fusion breakpoints is strongly limited. Indeed, the attempt to develop fusion detection tool based on WES data showed lower sensitivity compared to RNA-seq-based approach. 207
To detect structural rearrangements of chromosomes that lead to the formation of chimeric genes, a number of targeted DNA sequencing approaches have also been developed. Thus, the panel to detect both known and not yet described chimeric genes for regulatory kinases ALK, ROS1, RET, BRAF, MET, FGFR1-3, and NTRK1-3 has been developed. The panel consists of DNA probes labeled with biotin that are complementary to the intron and exon sequences of the genome, which are known breakpoints in the formation of chimeric genes. 208 Similar approaches have been used in commercial panels to search for chimeric genes from different manufacturers. Most fusions occur in intron sequences, which often contain repetitive sequences, such as insertions of transposable element, and are sequenced less efficiently than coding sequences. 209 This leads to false-negative results when the presence of the chimeric gene is confirmed by another method, for example, FISH or RNA sequencing.
Davies et al. compared three approaches for detecting rearrangements of ROS1 gene in lung cancer. 210 Targeted DNA sequencing did not detect 4 out of 18 chimeric genes confirmed by alternative approaches. The authors attribute false-negative results to specific structural features of specific introns.
Also, Benayed et al. analyzed the quality of chimeric genes detection using DNA sequencing panels. 211 14% of tumors with confirmed chimeric genes for clinically relevant tyrosine kinases were not detected when analyzed by the US FDA approved MSK-IMPACT panel, which is based on biotinylated oligonucleotides to capture genomic sequences of interest. False-negative results are also associated with the structural features of introns.
Sequencing of circulating cell-free DNA
Sequencing of circulating cell-free DNA from plasma of cancer patients revealed the presence of mutations characteristic for tumor DNA. 212 Sequencing of cell-free DNA from plasma of NSCLC patients with ALK mutation revealed acquisition of additional mutations in ALK and co-occurring amplification of MET1, 162 KRAS amplification, and a PI3KCA E545K mutation.213,214 This method requires prompt isolation of plasma for DNA extraction and permits non-invasive and cheap monitoring of cancer patients.
RNA sequencing
RNA sequencing is a method that has several advantages over DNA-based approaches. This approach has been proven informative for analyzing the expression level of various genes associated with the effectiveness of the response to anticancer drugs, 215 activation or inhibition of various molecular pathways.202,216,217 Approaches for the analysis of the mutational load of tumors according to full transcriptome analysis are also described.197,218 Therefore, the use of RNA sequencing makes it possible to assess the manifestation of various clinically important biomarkers in one experimental procedure.
The detection of chimeric transcripts has a number of advantages over DNA sequencing approaches. For example, when analyzing transcriptomic profile data, only transcriptionally active chimeric genes are identified, thereby filtering in only those fusions that might be the drivers of cancer progression and leaving out the passenger mutations. Second, in such a way both parts of a fusion gene can be identified at once – equally effective for both known and previously unknown fusions. Third, integrity of an open reading frame can be easily assessed, as well as the presence of a kinase domain in the chimeric gene product.
Targeted RNA sequencing
A variety of targeted panels have been proposed to search for both known and novel chimeric transcripts using RNA sequencing. Two approaches are widely used: selection of genes of interest by hybridization of cDNA with oligonucleotides labeled with biotin and complementary to exons of the target genes; or enrichment of libraries by PCR with specific primers complementary to the exon boundaries of the genes of interest and the universal adapter sequence (Figure 3).219,220

In a recent study, Heydt et al. compared four RNA sequencing and one DNA-sequencing-based targeted panels for the detection of chimeric genes from cell lines and FFPE tumor samples. 208 As in the studies described above, the DNA-based approach appeared to be less sensitive, producing more false negatives in the analysis of biopsies. Among the alternative RNA analytical panels, three were enriched by amplification: Archer FusionPlex Lung Panel (ArcherDX), QIAseq RNAscan Custom Panel (Qiagen), and Oncomine Focus Assay (Thermo Fisher Scientific). In turn, TruSight Tumor 170 Assay (Illumina) is based on hybridization of target sequences with biotinylated oligonucleotides. The best results were obtained for the TruSight Tumor 170 Assay (Illumina), which detected all chimeric transcripts in the samples under analysis. The single false-negative signal was observed in the results of ArcherDX and Qiagen panels; however, Qiagen panel returned more false-positive chimeras. The Thermo Fisher Scientific assay did not find 7 chimeric genes out of 18. The low sensitivity of the last panel is explained by the fact that the approach is based on classical PCR: both primers are complementary to known sequences of chimeric transcripts, which makes it impossible to detect chimeras that were never described previously and not included in the panel design. Thus, in general, RNA sequencing approaches are technically more effective than DNA analysis for the detection of chimeric oncogenes. 208
Methods for generation of sequencing libraries enriched in regions close to susceptible chimerization point are highly sensitive, especially for the detection of known structural rearrangements. However, this approach is limited by the set of probes available in the targeted panels, which does not allow detecting in such a way rearrangements at previously unknown loci. Also, the use of panels limits the set of biomarkers that can be analyzed in one experimental procedure. These shortcomings are devoid of total RNA or mRNA sequencing.
Whole transcriptome RNA sequencing for detection of fusion genes
Genome-wide RNA sequencing evaluates mutations of the transcribed DNA in an unbiased manner. A number of approaches have been proposed for the detection of chimeric transcripts in the analysis of full-transcriptome data. There are two main directions to the analysis of the RNA sequencing data for the detection of chimeric transcripts: (i) alignment of reads per genome and search for those reads that map to different loci or (ii) initial assembly of reads into long transcripts followed by a search for chimeras that do not map entirely to one genome region (Figure 4). 222

Scheme of operation of algorithms based on alignment (left) and primary assembly (right) (according to Haas et al., 222 modified).
In search for chimeric transcripts, Haas and colleagues used both approaches to compare 23 analytic algorithms. 222 The authors showed that the sensitivity of methods based on primary alignment is higher than for the approaches based on assembly of reads. The best analytic tools identified were STAR-Fusion, Arriba, and STAR-SEQR. It is important to note that algorithms for searching of the chimeric reads have been developed primarily for the analysis of cell lines and fresh frozen biopsies.223,224 Most of the work devoted to assessing their quality was carried out on artificial data, or again data obtained from cell lines and fresh frozen biopsies.225,226 However, paraffin-embedded biopsies fixed in formalin are the most common type of biomaterial in clinical practice, which is stored in collections for a long time.227,228
However, RNA in paraffin blocks is more degraded due to the fixation and storage procedures, which can lead to more false-negative results when searching for chimeric transcripts from the RNA sequencing data using existing algorithms. Validation of the work of the studied algorithms for this type of biomaterial has not yet been published.
Analysis of gene fusions from the FFPE material by RNA-seq
The use of DNA and RNA from archival material for genome-wide studies attract researchers’ attention for a long time and it was initially demonstrated that about 80% of genes, expressed in the fresh tissue can be detected in the FFPE samples by microarrays.229,230 Since then, we have witnessed considerable progress in the field and more recent investigations of RNA from paraffin blocks for gene expression analysis using microarrays 231 and sequencing228,232–234 revealed sufficient quality of RNA obtained from the FFPE samples to generate reproducible data consistent with RNA from the unfixed material. Furthermore, specific features such as conservation of open reading frame in both fusion partners, presence of RTK domain, and finding of several non-duplicate transcript reads for a fusion were shown as the efficient criteria for discriminating true versus artifact fusion reads in FFPE-derived RNA sequencing data. 235
Indeed, despite the absolute gene expression levels being not necessarily the same, very similar pathways were overrepresented within dysregulated genes obtained from the FFPE and freshly extracted RNA,232,233 thus demonstrating consistency with IHC studies.236,237
Fusion transcript detection by RNA-seq from FFPE samples was recently reviewed, focusing on the experimental variables; however, the difference in bioinformatics approaches of fusion transcript detention was not discussed. 238 Gene fusions were detected by RNA sequencing in 7 out of 8 cases of DNA-fusion positive fibrous histiocytomas. 239 Interestingly, comparison of ChimeraScan with TopHat software used in this study revealed better sensitivity of the former (9 versus 5 fusion transcripts detected) suggesting that detailed analysis of the software applications for the fusion transcript detection is needed. 239 It was shown that RNA-seq from the FFPE clinical material detects fusions with 94% (43 out of 46 fusions) concordance with DNA fusions, and one ST7-MET fusion was detected only by RNA-seq. 240
Analysis of single-cell RNA sequencing
High-throughput sequencing technology made single-cell RNA (scRNA) or DNA analysis possible at an unprecedented scale. Lately, several consortiums published aggregations and the analysis of the scRNA sequencing data.241–243
Functional enrichment analysis distinguishes different cell types as well as cancer cells, which also can be distinguished by the mutations and copy number variations typically observed in cancer, 244
Importantly, single-cell sequencing characterizes not only cancer cells, but also immune cells infiltrating the tumor. And, analysis of this data might reveal information which is relevant to tumor progression and treatment strategies. For example, sequencing of the T-cell receptor repertoire from glioblastomas treated by vaccination with heat shock protein peptide complex-96 identified dominant T-cell clones that reside in glioblastomas before treatment and stratify patients that are more sensitive to therapy. 245
Various methods have been developed to analyze gene expression of single cancer cells and to dissect their molecular subgroups. 246 Finding of gene fusions at single cell level can potentially shed light on specific features of cells and their subtypes. Several algorithms for fusion identification in bulk RNA-seq data have been developed,222,247 but detection of fusions on single cell level is still largely unsolved task. Indeed, the ambiguous and complicated library preparation steps result in generation of artificial chimeric reads and significant increase in the number of false-positive results. Moreover, the probability to detect fusion present in many cells is higher using bulk methods. Thus, several approaches both for sample preparation and for data analyses stages have been proposed. For example, using full-length scRNA-seq method enabled to detected and experimentally verify more gene fusions in scRNA-seq data than in bulk RNA-seq data for HeLa S3 cells. 248 These results were congruent with the finding published by another group of authors who could identify well-known as well as potential new fusion in colon cancer samples solely on single cell level. 249 However, experimental verification could not be performed here; thus, the increased number of fusions might at least partly represent artifacts of library preparation. 249 Another attempt to increase sensitivity of fusion detection at single cell level is to include specific primers targeted for the genes of interest. 250 This approach increased sensitivity of BCR-ABL1 detection in chronic myeloid leukemia cells. 250 However, this method is suitable only for the analysis of known fusions.
A more recent algorithm called scFusion was published for improving data analysis step. 251 This method utilizes both statistics and a deep learning model to exclude false-positive results. The algorithm also relies on the hypothesis that cells collected from one sample are more likely to contain the same gene fusions. 251 This limits its ability to detect rare or low expressed fusions, especially in highly heterogenous samples. Overall, scFusion requires greater sequencing depth as well as sequencing of larger amounts of cells from the same sample for obtaining reliable results. Analysis of the single-cell DNA sequencing of the NSCLC cohort enrolled in the MATCH-R (NCT0251782) trial that developed osimertinib resistance revealed heterogeneity of acquired mutations in the cells including FGFR3-TACC3, KIF5B-RE, and STRN-ALK fusions that can be treated by existing drugs, thus suggesting possible treatments to overcome osimertinib resistance. 252 Similarly, RNA sequencing revealed that ALK junctional heterogeneity in NSCLC may predict resistance to crizotinib. 253 Likewise scRNA-seq of chemoresistant cervical cancer revealed induction of the (PI3K)/AKT pathway. 254 Thus, it is possible to infer the mechanisms of acquired resistance and to monitor clonal changes of tumors in response to therapy.
Detection of fusions at single cell level can improve distinguishing cells subpopulations, thus shedding light on drug-resistant subclones in a tumor. However, this type of analysis still has strong limitations such as low coverage per individual cells, high PCR amplification bias and lack of standardization in data analysis.
Fusion oncogene databases
The fast growth of gene fusion data necessitates major organizational effort to gather them in the databases, and there is currently nearly a dozen of published databases of cancer fusion genes. Table 4 summarizes the common databases that are specified for fusion genes. One of the first databases designed to catalog gene fusions is the Mitelman database of chromosome aberrations and gene fusions in cancer that was first published in 1994. This database is supplemented with clinical association information that relate cytogenetic and genomic abnormalities, in particular gene fusions, to tumor characteristics or patient prognosis, based either on individual cases or associations. The database is searchable by a wide variety of fields, such as patient age, publication authors, gene, tumor histology, tissue type, mutation recurrence, associated clinical features, and cancer types. 255
Databases of gene fusions.

EST, expressed sequence tag; FISH, fluorescent in situ hybridization; TCGS, The Cancer Genome Atlas.
The Mitelman database was followed by COSMIC database in 2004, that started with only four genes, 273 then evolved to contain more than 5 million somatic mutations and more than 19 thousand gene fusions. 262
RNA-seq data is a major source of mining fusion transcripts. Several fusion databases were generated from transcript sequences available datasets, such as TCGA dataset. The Fusion Analysis Working Group identified 25 664 fusion events using RNA-seq data from tumor and normal samples from TCGA using multiple fusion calling tools. 223 Similarly, TumorFusions database is a searchable portal that catalogues over two thousand gene fusions detected in cancer and normal samples from TCGA.263,264
ChimerDB is another knowledgebase database of fusion genes265,274; this database contains fusions identified using bioinformatics analysis of transcript sequences compiled from GenBank and various other well-known public fusion databases and PubMed articles reporting fusion genes. ChimerDB is composed of three modules dealing with the analysis of deep sequencing data (ChimerSeq) and text mining of publications (ChimerPub) with extensive manual annotations (ChimerKB). 265
Newer gene fusion database is the ChiTaRS, generated by performing a bioinformatics analysis of transcript sequences and expressed sequence tags (ESTs) for multiple organisms in GenBank, starting from three organisms in the first version, 275 then extended to include eight organisms: human, mouse, fruit fly, rats, zebrafishes, cows, pigs, and yeast in the most recent version. 266 The latest version includes an extended information about fusions features, as well as 3D chromatin contact maps. In addition, FusionCancer database contains cancer fusion genes deduced from RNA-seq data. 267
Other databases combine fusion genes functional, regulatory, and genomic information. Example of such is the FARE-CAFE database that collect various aspects and data concerning fusion genes and proteins as protein domains, domain–domain interactions, protein–protein interactions, transcription factors and microRNAs. FARE-CAFE database incorporates chimeric transcripts and their Genomic information from different resources including Mitelman’s, dbCRID and the Trans location in Cancer (TICdb) databases. 268
dbCRID is a comprehensive database of human chromosomal rearrangements events and their associated diseases that documents the type of each event with the related disease or symptoms, the breakpoint positions and other genomic information. 271 Similarly, TICdb documents the precise location of each breakpoint inside a fusion gene. TICdb gather molecular information on gene fusions resulting from reciprocal translocation events associated with tumors. 272
Aside from cataloging gene fusions, numerous algorithms have been developed to predict fusion candidates from transcriptome data. Data aggregation and functional annotation with visualization support are necessary to assess the reliability, functional significance, and biological roles of predicted fusions. Programs such as GFusion, FusionScan, and STAR-Fusion are thought to be highly sensitive in detecting fusions with less false positives. 265 More recently, FusionHub introduced an integrated web platform that supports both annotation and visualization for the largest collection of fusion gene datasets aggregated from 24 resources. 276
HYBRIDdb is one of the earliest databases of hybrid genes to use a bioinformatics analysis for identifying gene fusions. HybridDB identified more than 3000 fusions from mRNA, EST, cDNA, and transcript sequences in the NCBI database. 269
Kim and Zhou built FusionGDB (Fusion Gene annotation DataBase) that gathers more than 40,000 fusion genes from fusion gene public resources such as TumorFusions and ChiTaRS 3.1. FusionGDB provides extensive functional annotations for these collected fusion events. Most importantly, the gene assessment across pan-cancer fusion genes, open reading frame (ORF) assignment and retention search of protein features. 277 FusionGDB was recently updated using deep learning techniques to provide eight categories of annotations: Fusion Gene Summary, Fusion Gene ORF analysis, Fusion Gene Genomic Features, Fusion Protein Features, Fusion Gene Sequence, Fusion Gene PPI analysis, Related Drugs, and Related Diseases. 270
Since kinase gene fusions are valuable biomarkers and promising drug targets, specific databases of such fusions have been developed. KuNG FU (KiNase Gene FUsion) is one of the largest curated databases, containing precise annotations on solely in-frame kinase gene fusions with intact kinase domain, which parameters were investigated in cancer cell lines. KuNG FU database is an available and informative tool for facilitating drug development and diagnostic studying. 270
Conclusions and further perspectives
Gene rearrangements stand among the major driver mutations in cancer. Recent developments unravel several therapeutically actionable fusion genes with approved targeted drugs effective against hematological and solid tumors. Many additional drugs targeting these and other fusion genes are in clinical trials and in development. Altogether, this gives physicians more options for the choice of therapy. However, cancer genomic instability leads to clonal heterogeneity and selection of cells with resistance to treatment, immune evasion, and metastatic potential, in many cases leading to failure of the therapy. Metastasis is the major problem in oncology. In particular, the picture becomes more complicated by the fact that each metastasis loci represents an individual clone with different mutational profile and drug sensitivity. To choose a proper therapy, it is critically important to provide a physician with actionable information about emerging clones. Thus, the future clinical oncology will utilize methods that will be able to measure mutations in the individual clones and assess the status of immune cells regulating tumor microenvironment. While single-cell sequencing methods that can measure clonal heterogeneity become available for academic research, targeted sequencing, FISH, and IHC remain the working horse of oncology.
To conclude, NGS-based methods provide several advantages for fusion detection both in clinical practice and in research studies. First, they combine fusion detection with finding of other clinically relevant biomarkers, thus providing more relevant information than ‘classical’ methods in just one test, which requires minute amounts of biosample. NGS approaches are suitable for the investigation of different sample types, including cell lines, fresh frozen, and FFPE biopsies. Different computational algorithms have been developed for the analyses of DNA and RNA NGS derived data and obtaining of highly reliable results. Further improvements in fusion detection approaches at both sample preparation and data analysis stages will expand the current knowledge of fusion frequencies among different cancer types, and of their particular impact on tumorigenesis, drug sensitivity, and resistance development. The latter has a strong potential of increasing efficacy of cancer treatment.