
Abstract
The discovery of activating mutations in epidermal growth factor receptor (EGFR) in non-small-cell lung cancer transformed the care and prognosis of patients and heralded the era of ‘personalized medicine’ in thoracic oncology. Osimertinib, a third-generation EGFR inhibitor, has been established as the preferred EGFR inhibitor for newly diagnosed patients which urged the need to develop treatment options for patients progressing on first-line osimertinib. However, acquired resistance invariably emerges and numerous efforts have been attempted to delay or overcome acquired resistance. In this article, we thoroughly reviewed the current understanding of osimertinib resistance mechanisms and explored the established and emerging treatment options. Newer treatment strategies targeting EGFR-dependent or -independent resistance mechanisms, novel approaches using bispecific antibodies and antibody–drug conjugates will be discussed. Moreover, what to do with brain only progression, and how to incorporate immunotherapy in EGFR-mutant lung cancer will be discussed. Lastly, future perspectives on the ongoing clinical trials and combination of front-line therapy will be introduced.
Introduction
The discovery of activating mutations in the epidermal growth factor receptor (EGFR) in a subset of lung adenocarcinomas in 2004 transformed the care and prognosis of patients with non-small-cell lung cancer (NSCLC) and heralded the era of ‘personalized medicine’ in thoracic oncology.1,2 EGFR mutations occur in about 15% of all lung adenocarcinomas, and are enriched among patients of younger age, minimal or no smoking history, and those of East Asian ethnicity. 3 While a variety of different EGFR mutations have been described in NSCLC, for the purposes of this review, we will focus on the two most common mutation subtypes, exon 19 deletions and L858R mutations in exon 21, which together comprise about 85% of all EGFR mutations in NSCLC.
Starting with the landmark iPASS study, 4 a series of randomized, phase III trials demonstrated improved progression-free survival (PFS) when first-generation EGFR tyrosine kinase inhibitors (TKIs; erlotinib and gefitinib) were compared to platinum-doublet chemotherapy in the first-line setting and established these drugs as a standard of care for newly diagnosed patients with EGFR-mutant NSCLC.4–7 Later studies also showed similar results with the second-generation EGFR inhibitor afatinib.8,9 While these agents all led to improvements in PFS over chemotherapy, long-term outcomes were limited by the development of acquired resistance, which was mediated by the EGFR T790M resistance mutation in ~50% of patients progressing on first- and second-generation EGFR inhibitors. 10
Third-generation EGFR inhibitors including osimertinib and others were developed to overcome T790M-mediated resistance and were found to be effective in T790M-positive patients following progression on erlotinib, gefitinib, and afatinib, leading to US FDA approval of osimertinib for this indication in 2015.11,12 Subsequently, the landmark FLAURA trial, which compared osimertinib to erlotinib and gefitinib as a first-line therapy for newly diagnosed patients with advanced EGFR-mutant NSCLC, demonstrated superior progression free (18.9 months) and overall survival (38.6 months) among patients treated with osimertinib.13,14 The results of the FLAURA trial established osimertinib as the preferred EGFR inhibitor for newly diagnosed patients, bringing urgency to the need to develop treatment options for patients progressing on first-line osimertinib. Figure 1 shows the timeline schematic of the approval of EGFR TKIs. In this review, we will summarize the current understanding of osimertinib resistance mechanisms and explore established and emerging treatment options.

Schematic timeline showing the approval status of EGFR TKIs.
Landscape of acquired resistance to osimertinib/third-generation EGFR TKI
Similar to what has been observed with acquired resistance to first- and second-generation EGFR inhibitors, resistance mechanisms to osimertinib can broadly be categorized as those mediated by alterations of EGFR itself (‘on-target’ resistance) and various ‘off-target’ resistance mechanisms including activation of parallel signaling pathways (‘bypass pathway activation’) and histologic transformations.15,16 While initial studies of osimertinib resistance focused on patients who received the drug in the T790M-positive setting,17,18 recent efforts focused on patients treated with first-line osimertinib. Emerging data from the FLAURA trial (which included matched pre- and post-treatment plasma from 91 patients who progressed on first-line osimertinib) and tissue biopsies obtained from 174 patients screening for the post-osimertinib ORCHARD study represent the largest cohorts of first-line osimertinib resistance reported to date and highlight several key findings which are summarized in Figure 2.19,20

Known resistance mechanisms to osimertinib after (a) first-line osimertinib treatment and (b) second-line or later-line osimertinib treatment.
First, on-target resistance, specifically secondary EGFR mutations, including C797X, G724S, and others, appear relatively uncommon among patients who receive osimertinib as their initial EGFR inhibitor. While prior studies had shown that up to 22% of treated with later-line osimertinib developed C797S, only 7% of patients developed C797S or other secondary EGFR mutations in the FLAURA and ORCHARD series. Amplification of EGFR was more commonly seen and frequently occurs in the absence of secondary EGFR mutations, but is not generally felt to be a targetable finding.
Taken together, ‘off-target’ resistance, including activation of parallel bypass tracks and/or downstream signaling pathways account for up to 30–50% of cases of first-line osimertinib resistance. Among these, MET amplification is the most common finding in the ORCHARD cohort, tissue NGS identified MET amplification in 24% of cases. Notably, in the cohort of patients with matched tissue and plasma testing, 53% cases of MET amplification were identified in tissue only, highlighting the limitations of liquid biopsies and circulating tumor DNA (ctDNA) analysis for the detection of MET amplification and likely accounting for the lower rate of MET amplification (15%) observed in the FLAURA ctDNA dataset. Acquired oncogene fusions have also been described but appear to be quite rare. For example, fusions in BRAF (5%), ALK (2%), and RET (<1%) were all observed in the ORCHARD matched tissue biopsy cohort. Finally, the ORCHARD dataset also identified mutations in the AKT/PTEN/PIK3CA pathway in 11% of patients, HER2 amplification or mutations in 2% and RAS mutations in <1%, while 10% of patients in the FLAURA ctDNA cohort had cell cycle gene alterations.
Importantly, neither the FLAURA nor ORCHARD cohorts included histologic assessment in their analysis of resistance mechanisms, a key limitation of both series. Data from Schoenfeld and colleagues suggest that histologic transformations occur in up to 15% of patients who progress on first-line osimertinib, with transformation from adenocarcinoma to small cell as well other non-small-cell (squamous, pleomorphic) histologies all seen in this context. As discussed below, patients with EGFR/RB1/TP53 co-mutations are at increased risk of SCLC transformation specifically.
In summary, the spectrum of resistance mechanisms to first-line osimertinib is broad with a diverse range of resistance mechanisms observed in both plasma and tissue biopsies. Despite recent advances, at least one-third of patients do not have an identified resistance mechanism, highlighting the need for further characterization of osimertinib resistance, including incorporation of methods to evaluate for non-genomic mechanisms of resistance which thus far have been limited.
For patients in the clinic today, evaluation for osimertinib resistance remains important in determining optimal next-line therapy. We recommend tissue biopsy and next-generation sequencing of both DNA and RNA as the preferred method to comprehensively evaluate for all known resistance mechanisms to osimertinib. Liquid biopsy can be considered if tissue biopsy is not feasible, but given the emerging data which suggests that liquid biopsies are most useful for the detection of acquired resistance mutations, ctDNA should not be considered a reliable method of detection of amplification, fusions, or histologic transformation. Where possible, treatment options should be aimed at identified resistance mechanisms as will be discussed in the remainder of this review. Nevertheless, with the increasing diversity of emerging resistance and the significant proportion of patients who do not have a targetable resistance mechanism identified, treatment options which are effective for broader groups of patients, rather than those aimed at each individual type of resistance, are urgently needed and likely to have the broadest clinical impact.
Strategies to overcome osimertinib resistance
Targeting EGFR C797S
For patients who develop resistance mediated by EGFR C797S, a new, fourth generation of EGFR TKIs specifically designed to target this mutation is now entering the clinic. Several promising candidates, including EAI045, JBJ-04-125-02, BLU-945, BBT-176, BDTX-1535, and others, have been evaluated preclinically and many are now entering phase I studies.
EAI045 is an EGFR allosteric inhibitor, which was effective both in vitro and in vivo in EGFR-mutant models including those harboring the C797S mutation. 21 However, EAI045 was not effective as a single agent, but required combination with cetuximab, which induced wild-type EGFR-mediated skin toxicities.
JBJ-04-125-02 is a more potent allosteric EGFR inhibitor which specifically targets EGFR L858R and inhibits EGFR L858R/T790M/C797S signaling in vitro and in vivo. 22 Dual targeting of EGFR with JBJ-04-125-02 and osimertinib led to enhanced apoptosis and delayed the onset of resistance. However, the selective activity of JBJ-04-125-02 against L858R and not exon 19 deletion is a key limitation, with agents active against both mutations now showing more promise.
BLU-945 is an investigational, reversible, highly wild-type selective next-generation EGFR TKI optimized for combination with other agents while targeting both activating and on-target resistance EGFR mutations, including T790M and C797S. BLU-945 demonstrated in vitro, in vivo antitumor monotherapy activity and enhanced activity in combination with osimertinib in resistant NSCLC models.23,24 Patient-derived xenograft models consistently showed that single-agent BLU-945 could lead to significant tumor growth inhibition in Ex19del/T790M/C797S mutation harboring model. Based on these preclinical results, a phase I/II study of BLU-945 in patients with metastatic EGFR-mutant NSCLC as monotherapy or in combination with osimertinib is ongoing (SYMPHONY; NCT04862780). Increasing BLU-945 doses have been associated with increasing antitumor activity with tumor shrinkage at doses of 200 QD and above, and one unconfirmed partial response seen at 400 mg QD. BLU-945 treatment also resulted in substantial decreases in ctDNA, with a reduction of 83% (10/12) and 82% (9/11) in EGFR T790M and EGFR C797S variant alleles, respectively. Dose-dependent tumor shrinkage and ctDNA variant allele fraction reduction were seen. 25
BLU-701 is also a reversible, selective EGFR inhibitor with nanomolar inhibitory activity against EGFR double mutations (Ex19del/C797S and L858R/C797S). 26 BLU-701 showed strong inhibition of activating EGFR mutations and double mutations, while sparing wild-type EGFR and showed significant brain penetration in in vivo models. BLU-701 is currently under clinical investigation in the ongoing HARMONY trial (NCT05153408).
BBT-176 showed an inhibitory effect on Ex19del/T790M/C797S and L858R/T790M/C797S triple mutations in engineered cell lines and patient-derived cell line and xenograft models. 27 A phase I/II trial of BBT-176 is ongoing to determine MTD and RP2D (NCT04820023). As of March 2022, early clinical data showed that steady-state drug exposure was proportional to dose and the drug’s long half-life was compatible with daily drug administration. No dose limiting toxicities (DLTs) were observed at dose levels up to 320 mg QD. Common treatment-related adverse events (TRAEs) were nausea (n = 5), vomiting (n = 3), diarrhea (n = 3), rash (n = 4), pruritus (n = 2), amylase increase (n = 2), and lipase increase (n = 2). No discontinuation of treatment due to TRAE has been reported so far. Reduction in EGFR mutation allelic frequency was observed in three patients, including a non-classical exon 19 deletion and T790M. These changes were correlated with tumor shrinkage in two of the patients. 28 Two patients with Ex19del/T790M/C797S showed radiological improvements in both target and non-target lesions.
Finally, JIN-A02 is a novel, orally available, fourth-generation EGFR TKI targeting C797S mutation and has demonstrated potent anti-tumor activity in preclinical models of double- or triple-mutant EGFR (ex19del/T790M or ex19del/T790M/C797S). 29 Its activity against a broad range of EGFR mutations, including L718Q which there is currently no treatment alternative, is expected to provide a therapeutic opportunity for patients who progressed upon previous EGFR TKI, and a future first-in-human trial is planned for testing clinical efficacy and safety.
Targeting MET amplification
MET amplification occurs in 10–25% of patients with EGFR TKI resistance,17,30 and may be overcome with combined EGFR and MET TKIs. 31 Recently, phase I safety data for the combination of savolitinib, a potent and selective MET TKI, in combination with osimertinib was reported from the global expansion cohorts of the TATTON trial. 32 These data suggested the feasibility of combining osimertinib and savolitinib as a new therapeutic option. The ongoing SAVANNAH study (NCT03778229) is evaluating the efficacy and safety of osimertinib and savolitinib in patients with MET-mediated resistance to osimertinib, with a phase III, randomized study of osimertinib/savolitinib versus platinum-doublet chemotherapy planned in the same patient population (SAFFRON; NCT05261399). In addition to savolitinib, anectodal responses to EGFR TKIs in combination with other MET inhibitors including crizotinib and capmatinib have also been reported.33,34
In addition to MET TKIs, additional strategies to target MET including both antibodies and antibody–drug conjugates (ADCs) are being developed. For example, REGN5093, which targets two different epitopes of MET, is being investigated in a first-in-human trial of MET-altered NSCLC (NCT04077099). REGN5093 has previously shown in vivo efficacy in MET-driven tumor models. 35 Telisotuzumab Vedotin (Teliso-V), an anti-MET ADC, has also shown favorable antitumor activity in a phase II trial in patients with NSCLC and c-MET protein overexpression. In an interim analysis, the EGFR wild-type group (n = 37) had an objective response rate (ORR) of 35.1% [95% confidence interval (CI), 20.2–52.5%] by independent central review (ICR) and an investigator assessed ORR of 36.1% (95% CI, 20.8–53.8%). Within this cohort, the median duration of response (mDOR) for patients in the c-Met intermediate subgroup was 6.9 months. However, the ORR in the EGFR-mutant group (n = 30) was only 13.3% (95% CI, 3.8–30.7%) by ICR and 25.8% (95% CI, 11.9–44.6%) when assessed by investigators. 36 In January 2022, US FDA granted breakthrough therapy designation for Teliso-V in EGFR wild-type NSCLC with high levels of MET overexpression.
Targeting oncogene fusions
For patients with acquired oncogene fusions, including ALK, BRAF, RET, and others, combination treatment strategies targeting EGFR and the relevant fusion can be considered. The coexistence of acquired RET rearrangement (CCDC6-RET) was first reported in 2015 in a case series, 37 in which the RET rearrangement was identified as a potential resistance mechanism to EGFR TKI-treated NSCLC patients. Later, a more comprehensive description of fusion-positive EGFR-mutant NSCLCs was reported, in which treatment with combination of RET and EGFR inhibitor was effective and well tolerated. Currently known rearrangements include RET (CCDC6-RET, NCOA-RET, ERC1-RET), BRAF (PCBP2-BRAF, AGK-BRAF, ESYT2-BRAF), ALK (EML4-ALK), FGFR3 (FGFR3-TACC3), NTRK1 (NTRK1-TPM3), and ROS1 (GOPC-ROS1). The most common rearrangements involve RET (43%) followed by ALK (26%), NTRK1 (16%), and FGFR3 (11%). 38
Combination of EGFR inhibitors and fusion inhibitors have shown early clinical responses as in the case of osimertinib and alectinib, and osimertinib and pralsetinib. 38 The ongoing post-osimertinib platform study, ORCHARD, will include RET and ALK fusion-positive cohorts to prospectively validate whether co-targeting EGFR and acquired RET and ALK fusions is efficacious and safe (NCT03944772).
Combination approaches involving bispecific antibody (amivantamab)
Amivantamab (JNJ-61186372) is a fully humanized bispecific antibody targeting EGFR and MET with several mechanisms of action.39,40 Amivantamab inhibits ligand-induced phosphorylation of EGFR and c-MET, induces antibody-dependent cellular cytotoxicity (ADCC) activity, and downregulates receptor expression in tumor cells. 41 Amivantamab was recently approved in the treatment of patients with advanced NSCLC with EGFR exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy. 42
CHRYSALIS is a phase I study of amivantamab, administered as a single agent and in combination with lazertinib, a third-generation EGFR TKI, to patients with advanced NSCLC harboring diverse EGFR mutations.43,44 In a cohort of patients with osimertinib-resistant EGFR-exon 19 deletion and L858R-mutant NSCLC treated with amivantamab monotherapy (n = 121) and amivantamab combined with lazertinib (n = 45), the ORR was 19% (95% CI, 12–27) and 36% (95% CI, 22–51), the mDOR was 5.9 (95% CI, 4.2–12.6) months and 9.6 (95% CI, 5.3–not reported) months, and the mPFS was 4.2 months and 4.6 months, respectively. 45 The improved response rate and durability observed with the amivantamab and lazertinib combination support the concept of dual targeting of both the extracellular and catalytic domains of EGFR. Infusion-related reactions were the most common adverse event (AE; seen in 69% of patients treated with amivantamab alone and 78% of patients treated with amivantamab/lazertinib combination), while other toxicities included paronychia (37% and 49%), acneiform dermatitis (28% and 51%), rash (26% and 27%), pruritus (22% and 31%), and hypoalbuminemia (26% and 38%). Of note, pneumonitis was reported in 2% and 4% of patients, respectively. 45
Responses to amivantamab/lazertinib were more common among patients with EGFR/MET-based resistance mechanisms to osimertinib (n = 17) where the ORR was 47% versus ORR 29% in patients without identified EGFR/MET-based resistance (n = 28). 46 In an exploratory analysis, EGFR/MET expression by immunohistochemistry was also correlated with response, with responses seen in 9/10 IHC-positive (defined as a combined EGFR + MET H score ⩾ 400) patients. These data suggest that the presence of EGFR/MET-mediated resistance mechanisms or EGFR/MET protein expression may be potential biomarkers of amivantamab/lazertinib response, but larger confirmatory studies will be needed to guide clinical use.
The CHRYSALIS-2 study also explored to combination of amivantamab/lazertinib in a larger cohort of patients EGFR-mutant NSCLC following osimertinib and platinum-based chemotherapy. In preliminary results, the ORR was 41% (95% CI, 24–61). In a separate cohort of heavily pre-treated patients (including 70% of patients who had received at least four prior lines of therapy), the ORR was 21% (95% CI, 11–36). 45 The ORR in patients in CHRYSALIS-2 was similar to patients post-osimertinib but chemo-naïve, suggesting intervening chemotherapy did not adversely impact on the activity of amivantamab and lazertinib. However, given the limitations of small sample size and cross-trial comparisons, these findings should be interpreted with caution.
Antibody–drug conjugates
Patritumab deruxtecan (U3-1402, HER3-DXd) is a novel HER3-targeted ADC composed of a fully humanized IgG1 monoclonal antibody to HER3 linked to deruxtecan, a topoisomerase I inhibitor payload via a tetrapeptide-based cleavable linker. 47 HER3 is overexpressed in 42–83% of NSCLC48,49 and is associated with poor prognosis. 50 In a multi-cohort phase I study of patients with EGFR-mutated NSCLC adenocarcinoma after failure of osimertinib and chemotherapy treated with patritumab deruxtecan at the recommended dose for expansion (5.6 mg/kg IV Q3W), the ORR was 39%. The median DOR and the median PFS were 7.0 months and 8.2 months, respectively. Similar outcomes were reported in the cohort of patients post-EGFR TKI ± platinum-based chemotherapy. 51 The activity of patritumab deruxtecan is highly encouraging in this heavily pre-treated patient population with a median of four prior lines of treatment, and importantly, patritumab deruxtecan was active in patients with diverse mechanisms of osimertinib resistance.
Datopotamab deruxtecan (Dato-DXd) is an ADC composed of a humanized anti-TROP2 (trophoblast cell-surface antigen 2) monoclonal antibody with the same payload (deruxtecan) and tetrapeptide-based cleavable linker. 52 TROP2 is highly expressed in NSCLC, regardless of genomic mutation status, and is also associated with a poor prognosis.53,54 Preliminary results from a phase I TROPION-PanTumor01 trial of patients with heavily pre-treated advanced NSCLC and actionable genomic alterations including 85% with EGFR mutations (69% with prior osimertinib) were recently presented. Among 34 patients treated with Dato-DXd at doses of 4–8 mg/kg q3w, the ORR was 35% and median DOR was 9.5 months. The most common AEs were nausea (62%) and stomatitis (56%). 55
Small-cell transformation, squamous cell transformation
While most EGFR-mutant lung cancers are initially diagnosed as adenocarcinoma, histologic transformations are a well-described resistance mechanism to all generations of EGFR inhibitors. Transformation to small-cell lung cancer (SCLC) was observed in 3–10% of patients who progress on first-generation EGFR inhibitors.10,56 More recently, Schoenfeld and colleagues identified histologic transformation in 15% of patients with tissue biopsies after osimertinib, and surprisingly showed that, in addition to SCLC, other histologies including both squamous and pleomorphic carcinomas could emerge at the time of resistance. 57 The overall frequency of histologic transformation after first-line osimertinib is poorly understood, as many of the series reported to date have been limited to the assessment of genomic resistance mechanisms through plasma and tissue.
The biology of SCLC transformation remains an area of active investigation. Early clinical and pre-clinical studies of patient-derived tumor samples and cell line models showed that RB1 loss, a hallmark of de novo SCLC, is nearly universal in transformed SCLCs and that, while activating EGFR mutations are retained at the DNA level, EGFR expression is largely lost following transformation.58,59 Subsequently, whole exome sequencing EGFR-mutant NSCLCs before and after transformation identified early inactivation of TP53 and RB1 as key risk factors for eventual SCLC transformation. Patients with EGFR/TP53/RB1 co-mutated lung adenocarcinomas had a 43-fold greater risk of transformation than those without these mutations. 60 Recently, this finding was validated in a clinical cohort of 39 patients EGFR/RB1/TP53-mutant lung cancers, wherein 7 (18%) of patients underwent eventual transformation. 61
While there are currently no clinical guidelines for the optimal management of patients with EGFR/RB1/TP53-mutant NSCLC, it is important to recognize the increased risk of SCLC transformation in this population, monitor closely for progression and obtain a tissue biopsy at the time of progression to evaluate for histologic transformation. For patients who undergo transformation, platinum-etoposide chemotherapy is the preferred treatment regimen. 59 The role of chemoimmunotherapy combinations now commonly used in de novo SCLC is unclear, though retrospective data suggest that immunotherapy given as monotherapy has little benefit. 59 Importantly, serial biopsies can identify re-emergence of NSCLC clones after transformation in some cases; thus, it is important to consider repeat biopsy if feasible for patients who experience disease progression after SCLC-directed therapy. 59
While histologic transformation from NSCLC to SCLC has been well described, shifts from adenocarcinoma to other non-small-cell histologies have also been observed and remain poorly understood. At present, there are no genomic markers of risk for squamous or other types of transformation, but recent data suggest that squamous histology can coexist with other known resistance mechanisms (including MET amplification, T790M, and others), highlighting the importance of complete molecular profiling, even in tumors where histologic transformation is identified. 62 For patients with transformation, chemotherapy regimens should be selected based on the predominant histology at the time of progression.
Brain progression on osimertinib, and what to do?
The patterns of disease progression after osimertinib can be largely categorized into three scenarios: isolated central nervous system (CNS) progression, systemic oligoprogression, and disseminated systemic progression. Oligoprogression is a relatively new concept which has emerged as effective local therapies became available, referring to progressive lesions restricted in both number and involved sites, typically less than five.63,64
Several real-world datasets have explored the patterns of disease progression after osimertinib. One report has suggested that EGFR T790M-positive NSCLC patients treated with osimertinib are more likely to progress with oligoprogressive lesions (72%) than overt systemic progressive lesions (28%). 64 Although the most common site of progression was lung, 15–20% of patients had disease progression in the brain.64–66 Furthermore, a majority of patients who had progression in the brain had oligoprogression. Notably, more than 60% of patients with oligo-progression continued osimertinib beyond progression with a median post-progression treatment duration of 4.1 months (some of them received a concurrent local ablative therapy, in most cases, radiotherapy). Nevertheless, osimertinib failed to demonstrate a significant benefit in overall survival (continued osimertinib; 18.9 versus discontinued osimertinib after PD; 15.1 months, p = 0.802).64,66
Nonetheless, osimertinib is renowned for its superior CNS penetration compared to older EGFR TKIs. 67 For treatment-naïve patients, osimertinib demonstrated an intracranial ORR of 66–91% and intracranial DOR of 15.2 months [95% CI: 4.1–not calculable (NC)]. 68 For previously EGFR TKI-treated patients, intracranial ORR of 54–70%, intracranial DOR of 8.9 months (95% CI: 4.3–NC), and intracranial PFS of 11.7 months (10–NC) were reported.69,70
Based on osimertinib’s intracranial efficacy, as well as previous data from the BLOOM study demonstrating intracranial efficacy and safety of osimertinib administered at 160 mg daily, 71 a recent retrospective analysis explored outcomes with osimertinib dose-escalation among 105 patients who experienced CNS disease progression on standard-dose osimertinib (80 mg daily). 72 Among 24 patients who were treated with osimertinib dose-escalation alone (cohort A), the median duration of CNS disease control was 3.8 months (95% CI: 1.7–5.8), specifically 5.8 months (95% CI: 1.7–5.8) and 2.0 months (95% CI: 1.0–4.9) for those with leptomeningeal and parenchymal metastases, respectively. The median duration of CNS disease control was marginally improved to 5.1 months (95% CI: 3.1–6.5) for patients who received concurrent radiation or cytotoxic chemotherapy (cohort B).
Several retrospective studies have reported the benefit of continued osimertinib treatment beyond radiographic progression. The duration of benefit varied widely with a second PFS of 5.7–12.6 months and 6.4–15.5 months for those who received radiation and for those who did not, respectively.73,74 Nonetheless, data on CNS efficacy of osimertinib in patients who continued beyond the first radiographic progression are scarce. Further studies are warranted considering the retrospective and non-comparative nature of study results reported so far.
Some clinicians aimed to restrict the disease progression by combining osimertinib with cytotoxic chemotherapy after progression to initial treatment. Frustratingly, a meaningful benefit was not demonstrated with combination therapy, with no new AEs. 75 The median PFS with osimertinib plus platinum-doublet regimen was 6.1–6.9 months.75,76 Randomized, placebo-controlled, prospective studies are ongoing to evaluate the efficacy of continuing osimertinib in combination with platinum-based chemotherapy in EGFR-mutant NSCLC patients harboring brain metastases who developed systemic disease progression but stable CNS disease (TORG1938/EPONA and COMPEL study).77,78
We must remember that a similar combination approach using early-generation EGFR TKI led to disappointing outcomes. The randomized, phase III, global IMPRESS trial recruited patients who had disease progression on first-line gefitinib and randomized them to receive platinum-doublet chemotherapy alone versus platinum-doublet chemotherapy plus continued gefitinib. 79 The median PFS did not differ between the two groups (5.4 months in both groups) but the median OS was detrimental with combination therapy [13.4 versus 19.5 months; hazard ratio (HR): 1.44, 95% CI: 1.07–1.94].79,80 Nevertheless, one of the main reasons for continuing osimertinib beyond the first radiographic progression is its CNS efficacy, unlike the IMPRESS study where main theoretical concern was the tumor flare after gefitinib discontinuation. Thus, the results from aforementioned prospective, randomized trials with osimertinib are highly anticipated.
In short, in the case of isolated CNS progression and oligo-progression, continuing osimertinib, with or without local radiotherapy, seems to be a valid option with limited additional benefit of osimertinib dose-escalation.81,82 Future studies will provide a clue on the continuation of osimertinib with or without cytotoxic chemotherapy in patients who experience disseminated systemic progression.
Combination with immunotherapy
The lack of benefit from immune checkpoint inhibitor (ICI) monotherapy in patients with EGFR-mutant NSCLC patients has been well documented,83–86 although the mechanism behind this phenomenon remains unclear. It is possibly related to the immune-suppressive tumor microenvironment in EGFR-mutant tumors.87,88 One theory suggests that the overexpression of CD39/CD73 in EGFR-mutant NSCLC inhibits the activity of various immune cells (such as CD8+ T cells, natural killer cells, and dendritic cells) by increasing extracellular adenosine level. 89
The inferior response of EGFR-mutant NSCLC to ICI monotherapy led to attempts to combine ICI with osimertinib.90,91 Nonetheless, this combination has been halted due to increased incidence of AEs, especially pneumonitis. 90 The TATTON trial which evaluated the combination of osimertinib with durvalumab reported a high incidence (22%, 5/23) of any grade interstitial lung disease (ILD) including two patients with at least grade 3 AEs, which led to an early termination of patient enrollment. 90 An increased incidence of AE in EGFR TKI and ICI combination was not limited to osimertinib. Grade 3 liver enzyme elevations were reported in gefitinib plus durvalumab or pembrolizumab combinations (40–70%).92,93
Even so, this phenomenon reserves a further investigation as the CAURAL trial which evaluated the same combination of osimertinib with durvalumab did not report an increased incidence of ILD (only one out of 14 patients in combination arm reported grade 2 ILD). The reason for different risk of AEs remains unclear and needs to be elucidated.91,94
Indeed, atezolizumab plus bevacizumab, carboplatin, and paclitaxel (ABCP) has emerged as a potential new standard of care for patients with EGFR-mutant metastatic non-squamous NSCLC who have failed on TKI treatment, as demonstrated from the subgroup analyses from IMPOWER-150 trial. 95 EGFR-mutant patients treated with ABCP regimen demonstrated a significant improvement in OS (HR: 0.31, 95% CI: 0.11–0.83), PFS (HR: 0.61, 95% CI: 0.36–1.03), and ORR (71% versus 42%) compared to those treated with BCP regimen. The OS benefit was evident in patients who had previous EGFR TKI therapy with HR of 0.39 (95% CI: 0.14–1.07).
A combination of ICI with chemotherapy has modest, if any, benefit on the survival outcomes in patients with EGFR mutations, as implied in IMPOWER-130 (atezolizumab in combination with carboplatin plus nab-paclitaxel). 96 Hence, the addition of bevacizumab to ICI/chemotherapy combination is a viable strategy to optimize treatment responses in patients with EGFR-mutant NSCLC by reverting the immune-permissive tumor microenviroment (TME) and normalizing tumor vasculature. 97
Several studies are ongoing to evaluate different combinations of ICI and anti-VEGF agents, such as pembrolizumab plus ramucirumab (NCT04120454) or pembrolizumab plus lenvatinib (NCT04989322), in EGFR-mutant NSCLC patients who had failed prior TKI treatments. Strategies to combine radiation with immunotherapy, to increase tumor antigen presentation and enhance T-cell infiltration into tumors, 98 are also adopted in early-stage EGFR-mutant NSCLC patients under neoadjuvant setting (NCT05244213 and NCT05319574) or concurrent chemoradiation (NCT04013542). The results from these diverse combinations are warranted.
Conclusions and future perspectives
Precision oncology in the treatment of EGFR-mutant lung cancers have revolutionized the treatment paradigm with the success of highly selective and potent EGFR inhibitors. Third-generation osimertinib demonstrated high response rate and survival gain in EGFR-mutant NSCLC patients in both treatment-naïve and treatment-refractory conditions, with fewer side effects compared to early generation TKIs. However, the development of on-target and bypass resistance mechanisms emerged as a challenge in the contemporary management of EGFR-mutant NSCLC who had become resistant to osimertinib. The present review focused on the currently available data on the mechanism behind the acquired resistance and the efforts to overcome them using different classes of drugs.
Ongoing efforts are investigating various upfront combination approaches with osimertinib which can change our treatment paradigm in the near future (Table 1). WJOG9717L which combined osimertinib with bevacizumab reported no PFS benefit compared to osimertinib alone. 99 FLAURA2 has safely opened up its investigation by demonstrating a manageable safety and tolerability in patients treated with osimertinib in combination with pemetrexed and platinum doublet chemotherapy. 100 We have previously experienced in NEJ009 trial that gefitinib in combination with chemotherapy could prolong PFS compared to gefitinib alone, 101 while the OS benefit was not apparent. 102 The efficacy results from FLAURA2 are highly awaited to see if third-generation osimertinib could lead to different outcomes to those of first-generation TKI. Other novel approaches such as combining EGFR/MET bispecific antibody with third-generation EGFR TKI in treatment-naïve EGFR-mutant NSCLC (MARIPOSA-1) are also paving way for an improved efficacy. 103
Current clinical trials for osimertinib-based combination approaches.
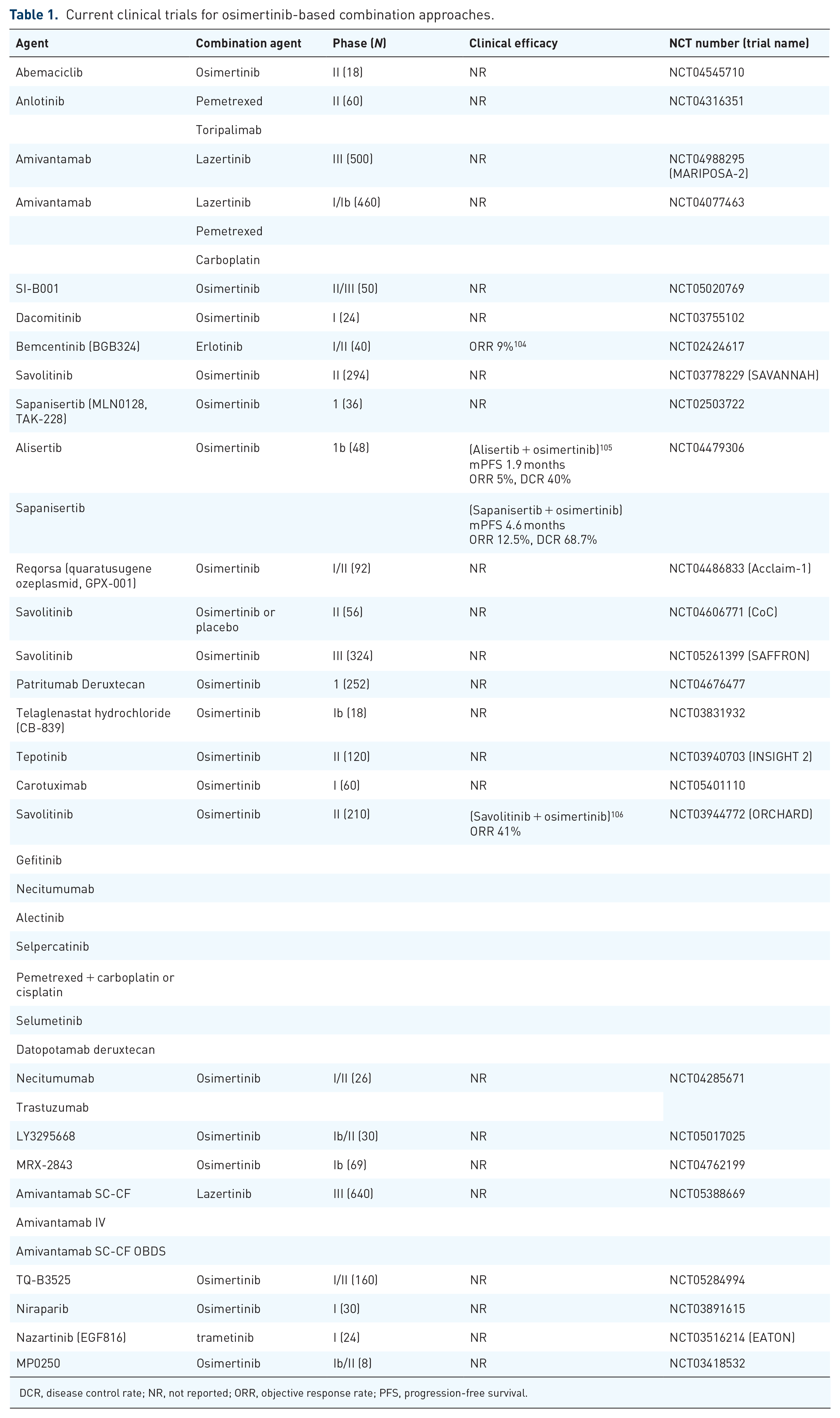
DCR, disease control rate; NR, not reported; ORR, objective response rate; PFS, progression-free survival.
Advanced research and clinical trials are necessary to overcome osimertinib resistance and improve the survival outcomes of EGFR-mutant NSCLC patients. In upcoming years, another paradigm-shifting breakthroughs in the treatment landscape of EGFR-mutant lung cancer patients is anticipated.