
Abstract
Background:
Electronic medical records (EMRs) have the highest value among real-world data (RWD). The aim of the present study was to propose a data collection framework of EMR-based RWD to evaluate the effectiveness and safety of cancer drugs by conducting a nationwide real-world study based on the Korean Cancer Study Group.
Methods:
We considered all patients who received ramucirumab plus paclitaxel (RAM/PTX) for gastric cancer and trastuzumab emtansine (T-DM1) for breast cancer at relevant institutions in South Korea. Standard operating procedures for systematic data collection were prospectively developed. Investigator reliability was evaluated using the concordance rate between the recommended input value for representative fictional cases and the input value of each investigator. Reliability of collected data was evaluated twice during the study period at three institutions randomly selected using the concordance rate between the previously collected data and data collected by an independent investigator. The reliability results of the investigators and collected data were used for revision of the electronic data capture system and site training.
Results:
Between the starting date of medical insurance coverage and December 2018, a total of 1063 patients at 56 institutions in the RAM/PTX cohort and 824 patients at 60 institutions in the T-DM1 cohort were included. Mean investigator reliability in the RAM/PTX and T-DM1 cohorts was 73.5% and 71.9%, respectively. Mean reliability of collected data in the RAM/PTX and T-DM1 cohort was 90.0% for both cohorts in the first analysis and 89.0% and 84.0% in the second analysis, respectively. Mean missing values of the RAM/PTX and T-DM1 cohorts at the time of simulation of fictional cases and final data analysis decreased from 20.7% to 0.46% and from 18.5% to 0.76%, respectively.
Conclusion:
This real-world study provides a framework that ensures relevance and reliability of EMR-based RWD for evaluating the effectiveness and safety of cancer drugs.
Introduction
Randomized controlled trials (RCTs) are typically considered the gold standard for generating clinical evidence. Patients enrolled in RCTs are highly selective populations subject to stringent eligibility criteria and managed in strictly controlled settings. This research format facilitates internal validity of the data obtained from RCTs to ensure the highest level of evidence for evaluating the efficacy and safety of new therapies prior to their implementation in clinical practice. However, these features of RCTs may contribute to a lack of external validity and generalizability to real-world clinical practice. It has been estimated that <5% of adult patients with cancer are enrolled in clinical trials. Therefore, it remains unclear whether the findings from RCTs are generalizable to the remaining >95% of the population and whether real-world patients in clinical practice respond to treatment in a manner similar to that in participants in RCTs.1–3 Therefore, real-world studies using data collected from diverse areas outside the scope of conventional RCTs may improve the generalizability and external validity of the data.4–6
In the framework for the United States Food and Drug Administration (FDA)’s Real-World Evidence Program, 7 real-world data (RWD), defined as data related to patient health status and/or the delivery of health care, are routinely obtained from medical sources. Examples of RWD include data derived from electronic health records, pharmacy claims and billing, product and disease registries, and patient-generated data (e.g. patient-reported outcomes and data from mobile or wearable devices). Real-world evidence (RWE) is defined as clinical evidence regarding the usage, risks, and benefits of a medical product derived from RWD analysis. 7 RWD and RWE have gained increased attention as complements to traditional clinical trials. The importance of RWE in oncology is also growing, considering that RCTs do not always account for the entire patient population that are candidates for specific cancer drugs.2,3 As cancer drug development is evolving rapidly, RWE is required to ensure drug effectiveness and safety.8–11 Recently, the FDA has accepted RWE to support approvals of original or supplementary indications for oncology drugs. RWE has added value to regulatory submissions for rare or orphan indications as a historical control (avelumab for Merkel cell carcinoma and blinatumomab for acute lymphoblastic leukemia), expanded access data (pembrolizumab for microsatellite instability-high or mismatch repair deficient cancers and lutetium Lu 177 dotatate for gastroenteropancreatic neuroendocrine tumors), and supported supplemental indication approval [palbociclib for male patients with breast cancer (BC)]. 12
Electronic medical records (EMRs) constitute RWD and include demographics, diagnoses, treatment decision-making, prescriptions, and laboratory or imaging data.13,14 Although EMRs enable the accumulation of large amounts of clinical data and are recognized as definitive data with the highest value among RWD, minimizing the biases that may affect study outcomes remains challenging. Commencement of real-world studies using EMR-based RWD requires study designs that minimize such biases, and data quality management is a key requirement for the extracted data in RWE.
Here, we conducted a nationwide real-world study and proposed a framework for systematically collecting EMR-based RWD to evaluate the effectiveness and safety of cancer drugs. We selected ramucirumab plus paclitaxel (RAM/PTX) and trastuzumab emtansine (T-DM1), which are currently used to treat advanced gastric cancer (GC) and BC, respectively, and systematically collected EMR-based RWD at relevant institutions in South Korea.
Patients and methods
The Korean Cancer Study Group (KCSG) conducted a nationwide real-world study to develop a data collection system for EMR-based RWD for cancer drugs. The consortium of experts for the study comprised oncologists, statisticians, and site management organization. The data collection process for EMR-based RWD is illustrated in Figure 1.
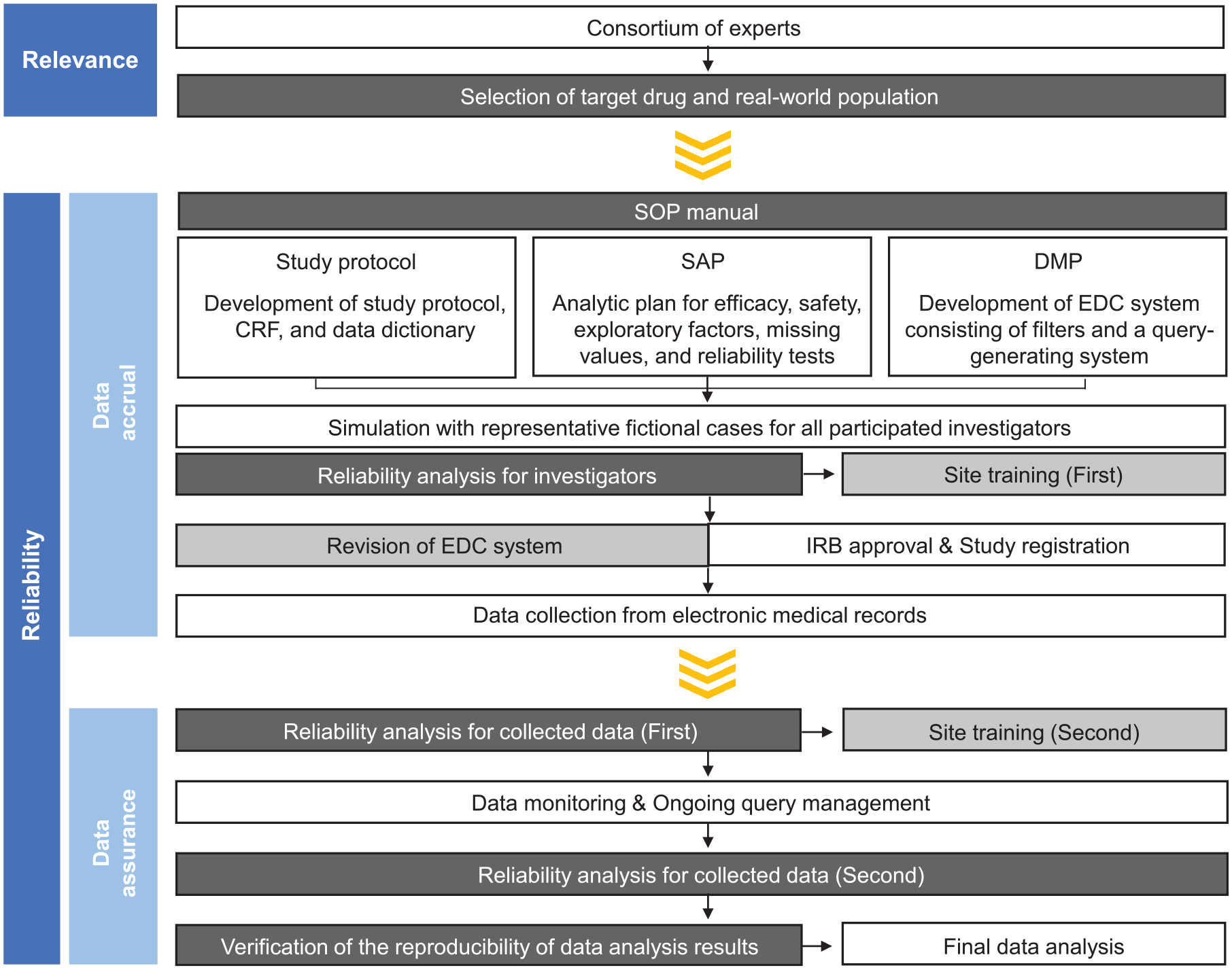
A framework for collection of EMR-based RWD.
Selection of real-world population
RAM/PTX for GC and T-DM1 for BC have been covered by medical insurance in South Korea since May 2018 and August 2017, respectively. We considered all patients who received each drug between the starting date of medical insurance coverage and December 2018 for enrollment in the study. We excluded patients treated at institutions that included less than two candidate patients, those without institutional review board (IRB) approval, or those who declined to participate. We used eligibility criteria similar to those in the pivotal RAINBOW RCT for RAM/PTX and EMILIA RCT for T-DM1.15,16 In the RAM/PTX cohort, patients with locally advanced unresectable or metastatic gastric or gastroesophageal junction adenocarcinoma, who experienced disease progression after first-line fluoropyrimidine and platinum-containing combination chemotherapy and received palliative second-line RAM/PTX treatment, were included. In the T-DM1 cohort, patients with human epidermal growth factor receptor 2-positive locally advanced unresectable or metastatic BC, who had previously been treated with trastuzumab and taxane and received palliative T-DM1 treatment were included.
This study was conducted by the GC and BC committees of the KCSG with funding from the Health Insurance Review & Assessment Service (HIRA). The study was performed in accordance with Good Clinical Practice guidelines and approved by the IRB of each hospital (Supplemental Tables 1 and 2). The IRBs of all participating institutions waived the requirement for informed consent from patients due to the retrospective nature of the study. This study is registered with ClinicalTrials.gov, with identifiers NCT04192734 (KCSG ST19-16) and NCT04202328 (KCSG BR19-15).
Standard operating procedure manual
To optimize the reliability of the RWD and minimize bias, standard operating procedures (SOPs) are required in retrospective studies using RWD. 7 We prospectively developed an SOP manual that included the study protocol, the common case report form (CRF), a dictionary to define common data elements, a statistical analytic plan (SAP), and a data management plan (DMP) (Supplemental materials).
Simulation with representative fictional cases and reliability analysis for investigators
We developed two representative fictional cases treated with each drug (Figure 2). The simulation for each case was performed by investigators to analyze the reliability of the participating investigators. Investigator reliability was evaluated using the concordance rate between the recommended input value for representative cases and the input value of each investigator. The inter-investigator agreement was evaluated using the ratio of matching pairs among all possible pairs between investigators. When n investigators evaluate the ith variable with m possible response values (categories), the agreement rate of the ith variable can be obtained as follows:

where

Simulation with representative fictional cases for each drug.
Data collection and reliability analysis for the collected data
All data were anonymously collected and managed using an electronic data capture (EDC) system consisting of filters and a query-generating system to guarantee reliability and control for errors and missing or inconsistent data. A reliability analysis for collected data was conducted twice during the study period at three institutions randomly selected by statisticians. The first analysis was performed for two institutions at the 50% data collection timepoint. The second analysis was performed for one institution at the completion of data collection. Five patients were randomly selected from each institution and an independent investigator, other than the investigator who collected CRF data from the selected institution, completed the CRF for each case. The reliability of collected data was evaluated using the concordance rate between the previously collected data and data collected by the independent investigator.
Verification of the reproducibility of data analysis results
Two statisticians independently analyzed the same data and compared their results to evaluate data analysis reproducibility. The statisticians only shared the study purpose, collected data, and analysis methods or modeling. If the analysis methods or results differed, the most appropriate method and results were determined by consensus with another statistician and oncologists.
Statistical analysis
During CRF development, ‘not applicable’, which does not require entry, was separately classified and coded from ‘missing’ values treated as blanks. Missing values were presented as separate data by calculating the missing rate for each variable.
All data are reported as mean and standard deviation (SD) or proportion (%). All analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
Real-world population
The participating institutions and patients are shown in Table 1. Between the starting date of medical insurance coverage and December 2018, a total of 1460 patients with GC at 92 hospitals and 1029 patients with BC at 87 hospitals received RAM/PTX and T-DM1, respectively, in South Korea. In the RAM/PTX cohort, 1336 patients at 57 institutions were assessed for eligibility and 1063 eligible patients at 56 institutions were included. In the T-DM1 cohort, 972 patients at 60 institutions were assessed for eligibility and 824 eligible patients were included in the analysis.
Participating institutions and patients.

IRB, institutional review board; RAM/PTX, ramucirumab plus paclitaxel; T-DM1, trastuzumab emtansine.
Reliability of participant investigators
Reliability of the participating investigators is presented as investigator reliability and inter-investigator agreement rates in Table 2. Reliability of the participant investigators by category for major CRF variables is presented in Figure 3. From the 56 participating institutions in the RAM/PTX cohort, 53 investigators (94.6%) (one per institution) participated in case simulation. The investigator reliability for all 268 variables and 34 major variables of the CRF were 60.5 ± 27.1% and 73.5 ± 21.3%, respectively (Table 2). The inter-investigator agreement rates including and excluding missing values were 65.0 ± 21.0% and 86.0 ± 16.0%, respectively (Table 2). CRF variables with investigator reliability and inter-investigator agreement excluding missing values <70% were related to baseline characteristics (age and date of diagnosis for locally advanced or metastatic disease) and adverse events (Figure 3(a) and Supplemental Table 3). CRF variables with a proportion of missing values >10% were associated with RAM/PTX treatment (discontinuation and reasons for discontinuation), adverse events, and survival (date of death or the last follow-up visit) (Figure 3(a) and Supplemental Table 3).
Reliability of participating investigators.

The concordance rate between the recommended input value for representative fictional cases and the input values of each investigator.
The ratio of matching pairs among all possible pairs between investigators.
CRF, case report form; RAM/PTX, ramucirumab plus paclitaxel; SD, standard deviation; T-DM1, trastuzumab emtansine.

Reliability of the participating investigators and inter-investigator agreement for major variables of case report form in the (a) ramucirumab plus paclitaxel and (b) trastuzumab emtansine cohorts. Each bar represents mean values of variables in each category.
From the 60 participating institutions in the T-DM1 cohort, 58 investigators (96.7%) (one per institution) participated in case simulation. The investigator reliability for all 152 variables and 28 major variables of the CRF were 66.4 ± 21.6% and 71.9 ± 16.9%, respectively (Table 2). The inter-investigator agreement rates including and excluding missing values were 61.0 ± 17.0% and 83.0 ± 19.0%, respectively (Table 2). CRF variables with investigator reliability and inter-investigator agreement excluding missing values <70% were associated with baseline characteristics (date of initial diagnosis of BC), previous systemic treatments (protocol), tumor response (date of best response), adverse events, post-discontinuation therapy (protocol), and survival (Figure 3(b) and Supplemental Table 4). CRF variables with a proportion of missing values >10% were related to baseline characteristics at initial diagnosis of BC, previous systemic treatments, central nervous metastasis, adverse events, and post-discontinuation therapy (protocol) (Figure 3(b) and Supplemental Table 4).
Reliability of the collected data
For each drug, previously collected data and the data collected by independent investigators were compared for 10 patients from two institutions in the primary analysis and five patients from one institution in the secondary analysis. In the RAM/PTX cohort, the concordance rates for the input values of all CRF variables and major CRF variables in the first analysis were 90.0 ± 20.0% and 90.0 ± 10.0%, respectively, and the concordance rates in the second analysis were 88.0 ± 23.0% and 89.0 ± 15.0%, respectively (Table 3). In the T-DM1 cohort, the concordance rates for the input values of all CRF variables and major CRF variables in the first analysis were both 90.0 ± 10.0%, and the concordance rates in the second analysis were 94.0 ± 12.0% and 84.0 ± 17.0%, respectively (Table 3). The input data discrepancies between the previously collected data and the data collected by independent investigators were due to the input of variables that were either in textual form or not directly described in the EMRs (performance status, reason for discontinuation, adverse events, best response or disease progression, and survival data), different input date criteria according to each investigator (e.g. variability in examination date, date of result report, date of result confirmation, or date of visit), or incorrect data input based on data collection dates other than the data cutoff date (Supplemental Tables 5 and 6).
Reliability of the collected data.

The concordance rate between the previously collected data and data collected by independent investigators.
CRF, case report form; RAM/PTX, ramucirumab plus paclitaxel; SD, standard deviation; T-DM1, trastuzumab emtansine.
Site training and changes in the missing values
First site training adopted the simulation results with representative fictional cases for each drug. Educational information for each institution was delivered by analyzing the variables that had low agreement with the recommended input values of representative cases or high incidence of missing values. EDC system consisting of filtering and a query-generating system were revised to reflect the CRF variables with low reliability of investigators or high incidence of missing values. Subsequent second site training adopted the first reliability results of the collected data conducted at two institutions. Educational information was delivered by analyzing the variables that did not match the input values of investigators at the selected institution and the input values of independent investigators in cross-checking.
Through revision of the EDC system and site training, the missing values of the RAM/PTX and T-DM1 cohorts at case simulation and final data analysis (mean ± SD) decreased from 20.7 ± 21.1% to 0.46 ± 0.59% and from 18.5 ± 13.7% to 0.76 ± 1.48%, respectively (Figure 4).
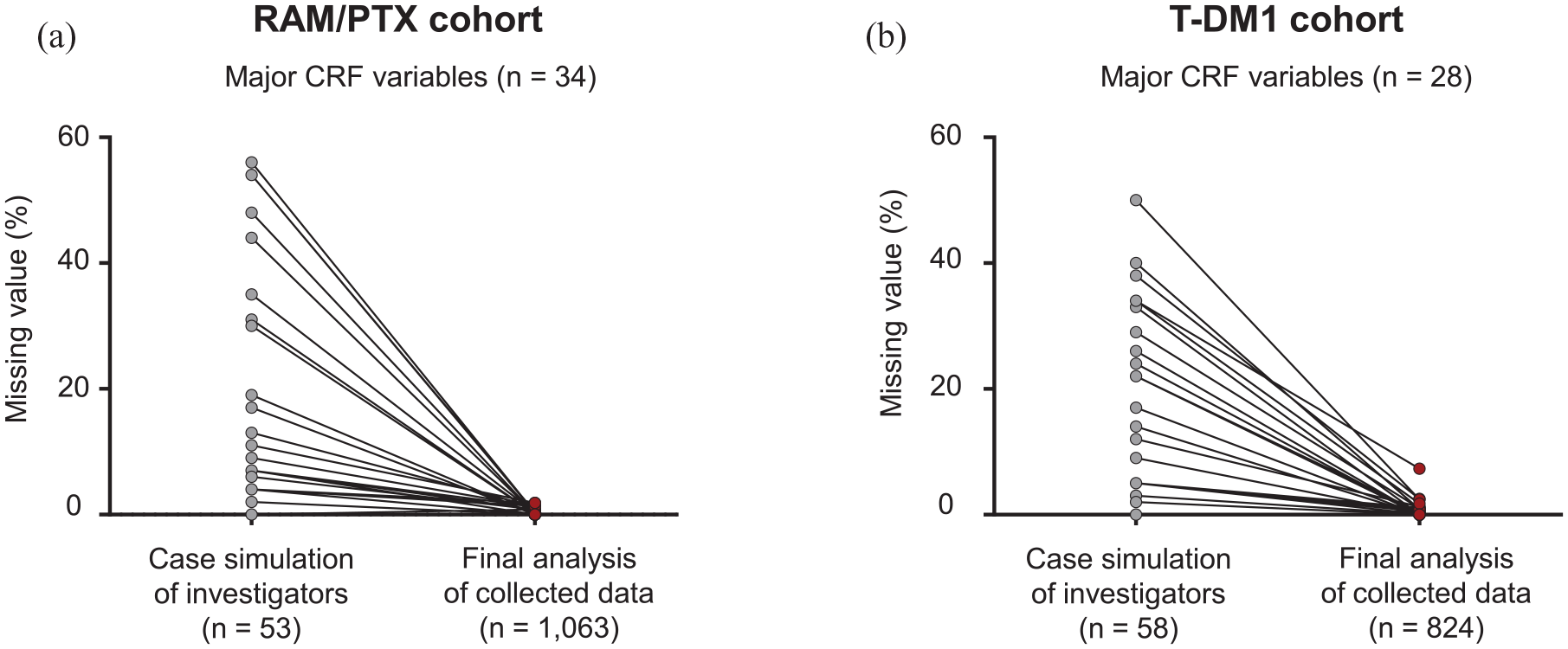
Changes in missing values for major variables of case report in the (a) ramucirumab plus paclitaxel and (b) trastuzumab emtansine cohorts.
Reproducibility of data analysis results
The analysis results were consistent between statisticians for most variables, but differences were observed in the total number of included patients, time to progression, and progression-free survival. For the included patients, one statistician included all patients with eligibility criteria of ‘yes’, while the second statistician included patients who met the eligibility criteria and for whom survival data were available (no missing values). For patients with both disease progression and treatment other than the investigational drug in the T-DM1 cohort, one statistician defined the time of disease progression as an event, while the second statistician defined the occurrence of other treatments as censoring. By consensus with a third statistician and oncologist, the included patients were defined as those who met the eligibility criteria with available survival data, an event was defined as disease progression or death, and censoring was defined as receiving other treatments or non-occurrence of events. After clarifying the definitions of included patients, events, and censoring, the results regarding the number of included patients, time to progression, and progression-free survival were consistent between the two statisticians.
Discussion
This large-scale, nationwide, real-world study provides a systematic framework for collecting EMR-based RWD with a definitive design and detailed methodologies for data quality management. To the best of our knowledge, no previous study has focused on a detailed data collection process for EMR-based RWD. Previous real-world studies were only based on retrospective data collection and analysis results or were conducted in selected populations.17–22 Our study is notable in that the systematic collection process for EMR-based RWD was prospectively planned and conducted to minimize bias and increase RWD reliability in nearly all the populations that received candidate drugs.
The KCSG ST19-16 and KCSG BR19-15 studies reported favorable effectiveness and safety of RAM/PTX 23 and T-DM1 and supported the scientific evidence derived from pivotal RCTs. The strengths of such RWE are dependent on the relevance of the underlying data and reliability of data accrual and data assurance (data quality control).7,24,25 In general, the FDA and other stakeholders assess the relevance and reliability of RWD to determine the suitability of RWD for regulatory decision-making. 7
The overall assessment of the relevance of RWD determines whether the existing RWD source is appropriate for evaluating drug effectiveness and safety. In the present study, we considered all patients who received RAM/PTX for GC and T-DM1 for BC during the study period and applied similar eligibility criteria to those used in pivotal RCTs that provided evidence for drug approval and reimbursement.15,16 Therefore, our study employed an all consecutive-comer’s design that minimized potential bias in patient selection and enrollment. Furthermore, the real-world population in our study was appropriate for evaluating drug effectiveness and safety.
The reliability of RWD is assessed by evaluating how the data were collected (data accrual) and whether the data collection and analysis procedures provide adequate assurance for error minimization and sufficiency of data quality and integrity (data assurance). In addition, standardization of uniform and systematic methods for collecting and cleaning data is vital for the reliability of RWD. To ensure RWD reliability, SOP for the use of uniform and systematic methods for data collection and analysis is essential to ensure data quality. The SOP manual in our study described the data elements to be collected, data element definitions, methods for data aggregation and documentation (e.g. CRF form, survey guidelines), SAP (e.g. analytic plan for efficacy and safety, exploratory factors, missing values, and the reliability of investigators and the collected data analyses), and DMP (e.g. EDC system consisting of filters and a query-generating system). In our study, RWD reliability was evaluated based on investigators and the collected data. To evaluate the reliability of investigators, we developed two representative fictional cases treated with each drug. Furthermore, the simulation results with representative fictional cases were used for revision of EDC system and site training. Notably, training all investigators at the participating sites is challenging because real-world studies involve a greater number of institutions and investigators than conventional trials. As such, simulation via representative fictional cases is an effective training method that prepares investigators for participation in real-world studies. Data assurance via data quality control is essential for providing confidence in the reliability of RWD.7,24,25 Reliability analysis for the collected data was conducted twice during the study period and evaluated using the concordance rate between previously collected data and data collected by independent investigators. Ongoing additional site training utilized the initial reliability results of the collected data that did not match the input values of investigators in selected institutions and the input values of independent investigators. All the collected data were managed using an EDC system consisting of filters and a query-generating system to control for errors and missing or inconsistent data. Although reliability of the participating investigators was not evaluated again at the time of final data analysis, the reliability of collected data was improved and the missing values were significantly reduced when compared with the extent at the time of case simulation based on the site training and EDC system. Furthermore, to ensure the reproducibility of data analysis results, two statisticians analyzed the same data independently and compared the analysis results. In this regard, RWD assurance can be improved by evaluating data reliability and consistency over time, implementing ongoing training and data monitoring by query-generating systems, and performing independent data analysis by different statisticians.
Despite meticulous data quality management in our data collection process, we identified several variables with low reliability in the second analysis of the collected data. When the data recorded in EMRs were not categorized and were in textual form (e.g. performance status, previous treatments, adverse events, reasons for discontinuation, best response or disease progression, and post-discontinuation therapy), the concordance of investigators was low. In this real-world study, we evaluated the format of the EMRs of patients with cancer receiving RAM/PTX at 55 institutions in South Korea (data not shown). Among CRF variables, approximately 50% of cancer-related or chemotherapy-related variables were described in a textual form in EMRs. The coding of major variables for RWD in the EMRs of cancer patients (e.g. performance status, tumor response, disease progression, and survival) will help to reduce missing data and improve the reliability of EMR-based RWD. Another limitation in the EMR-based RWD collection is that data cannot be collected in the EMRs of each hospital. Even death-related data, which are considered to be the key endpoint in cancer patients, did not exhibit 100% agreement between investigators because EMR data for patients whose death occurred outside the institution were unreliable. In our study, survival could not be accurately determined (indicated as ‘unknown’ in CRF) in 23.5% patients from the RAM/PTX cohort and 17.7% patients from the T-DM1 cohort. Therefore, it is necessary to link or integrate data with other public institutions that can determine the patient death information while protecting personal information to facilitate reliable RWD collection in cancer patients. Furthermore, there may be human error in our EMR-based RWD collection; big data management systems, such as Clinical Data Warehouse, could not be used because the EMR systems were inconsistent across institutions. Therefore, there is a need for a digital healthcare system herein EMR-based RWD can be structured, defined, formatted, and exchanged with an integrated computer system and converted into scientific data.
The data collection framework for EMR-based RWD proposed in the present study was based on the principles of the FDA’s RWE program (Figure 1). 7 The study population included nearly all cancer patients treated with candidate cancer drugs and was representative of routine clinical practice in South Korea. Furthermore, a detailed SOP manual for collection of uniform RWD and minimization of bias was developed prior to study initiation to ensure data quality. This framework included reliability tests for investigators and the collected data, site training programs, EDC system consisting of filtering and a query-generating system, and verification of the reproducibility of data analysis results. Therefore, the data collection process in our study provides a framework that ensures relevance and reliability of EMR-based RWD for evaluating the effectiveness and safety of cancer drugs. In the future, it will be necessary to develop a big data digital healthcare system that integrates this data collection process and cooperates with public institutions, such as the HIRA and the National Health Insurance Service.
The present framework has several limitations. First, it was inherently limited due to the retrospective observational methodology, including missing data, under-reporting of adverse events and deaths, and the lack of control patients, which could have influenced data analyses for drug effectiveness and safety. Prospective RWD collection would generate evidence based on pragmatic clinical trials that support randomized study designs and extend clinical research to the point of care.7,26,27 Second, EMR data may not capture all data elements required to answer the question of interest. In that regard, administrative claims may provide a beneficial overlap of information to investigate the balance between costs and benefits.7,28–32 In addition, novel technologies (e.g. mobile or wearable devices or applications) will enable data collection from candidate patients with cancer in real time. These sources contain vital health-related information, such as demographics, symptoms, long-term effects, adherence rates, and financial burden.7,33–36 Therefore, future research should focus on developing a data collection framework for RWD derived from prospective studies, pharmacy claims and billing, patient-generated data, and novel technologies. The KCSG has conducted a prospective study (KCSG ST21-06) to establish a RWD collection framework to evaluate not only drug effectiveness and safety, but also quality of life, patient-reported outcomes, financial burdens, and smartphone-based healthcare data (ClinicalTrials.gov with identifiers NCT04915807).
In conclusion, the KCSG has performed a nationwide real-world study and developed a data collection framework to ensure the relevance and reliability of EMR-based RWD for evaluating the effectiveness and safety of cancer drugs. The data collection framework developed here will be a valuable reference for establishing RWE by systematically collecting EMR-based RWD for cancer drugs regardless of the type of cancer and anticancer drugs.
Supplemental Material
sj-docx-1-tam-10.1177_17588359221132628 – Supplemental material for Data collection framework for electronic medical record-based real-world data to evaluate the effectiveness and safety of cancer drugs: a nationwide real-world study of the Korean Cancer Study Group
Supplemental material, sj-docx-1-tam-10.1177_17588359221132628 for Data collection framework for electronic medical record-based real-world data to evaluate the effectiveness and safety of cancer drugs: a nationwide real-world study of the Korean Cancer Study Group by Hye Sook Han, Kyoung Eun Lee, Young Ju Suh, Hee-Jung Jee, Bum Jun Kim, Hyeong Su Kim, Keun-Wook Lee, Min-Hee Ryu, Sun Kyung Baek, In Hae Park, Hee Kyung Ahn, Jae Ho Jeong, Min Hwan Kim, Dae Hyung Lee, Siheon Kim, Hyemi Moon, Serim Son, Ji-Hye Byun, Dong Sook Kim, Hyonggin An, Yeon Hee Park and Dae Young Zang in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
This work was supported by the Korean Cancer Study Group (KCSG) and the Health Insurance Review & Assessment Service (HIRA). The views expressed are those of the authors and not necessarily those of HIRA. We thank all investigators and their support staff who generously participated in this study.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.