
Abstract
Germline replication-repair deficient (gRRD) gliomas are exceptional events, and only a few of them have been treated with immune checkpoint inhibitors (ICIs). Contrary to sporadic gliomas, where ICIs have failed to show any objective benefit, the very few patients with gRRD gliomas treated with ICIs to date seem to benefit from programmed-death-1 (PD-1) inhibitors, such as nivolumab or pembrolizumab, either in terms of durable responses or in terms of survival. T-cell immunohistochemistry (IHC) and T-cell receptor (TCR) repertoire using high-throughput next-generation sequencing (NGS) with the Oncomine TCR-Beta-SR assay (Thermo Fisher Scientific) were analyzed in pre- and post-nivolumab tumor biopsies obtained from a patient with a Lynch syndrome-associated glioma due to a germline pathogenic hMLH1 mutation. The aim was to describe changes in the T-cell quantity and clonality after treatment with nivolumab to better understand the role of acquired immunity in gRRD gliomas. The patient showed a slow disease progression and overall survival of 10 months since the start of anti-PD-1 therapy with excellent tolerance. A very scant T-cell infiltrate was observed both at initial diagnosis and after four cycles of nivolumab. The drastic change observed in TCR clonality in the post-nivolumab biopsy may be explained by the highly spatial and temporal heterogeneity of glioblastomas. Despite the durable benefit from nivolumab, the scant T-cell infiltrate possibly explains the lack of objective response to anti-PD-1 therapy. The major change in TCR clonality observed after nivolumab possibly reflects the evolving molecular heterogeneity in a highly pre-treated disease. An in-deep review of the available literature regarding the role of ICIs in both sporadic and gRRD gliomas was conducted.
Introduction
Lynch syndrome (LS) is an autosomal-dominant inherited disease characterized by germline mutations in mismatch-repair (MMR) genes with an increased risk of colorectal, endometrial, ovarian, and urinary tract cancers. In addition, the occurrence of gliomas is recognized as a rare presentation of LS. The so-called Turcot syndrome refers to those patients with both colorectal cancer (CRC) and primary malignant central nervous system (CNS) tumors secondary to either LS or to familial adenomatous polyposis (APC). MMR-deficient (MMRd) malignancies secondary to LS are microsatellite instability-high (MSI-H) tumors, characterized by a high tumor mutational burden (TMB) that translates into a high neoantigen load that permits an easier and more straightforward recognition by the immune system, which may explain the higher objective responses and durable benefit seen with immune-checkpoint inhibitors (ICIs) in these tumors.1,2 While ICIs against the programmed-death-1 (PD-1)/programmed death-ligand 1 (PD-L1) axis have not shown efficacy either alone or in combination with bevacizumab or temozolomide in sporadic newly diagnosed or recurrent glioblastoma (GB) (Table 1), a few reported cases of GB associated with germline replication-repair defects (gRRDs) have shown tumor responses to anti-PD-1 agents.3–20 Brilliant responses to these agents have been reported in biallelic mismatch-repair deficiency syndrome (BMMRDS) characterized by the appearance of CNS tumors and hematological cancers at early ages. Benefit from ICIs has also been reported in LS-associated GB (Table 2). Contrary to these gRRD-driven tumors, most patients with somatic replication-repair deficient (RRD) high-grade gliomas (HGG) have not been shown to derive benefit from ICIs17,21–23 (Table 2). We report the case of a patient with LS due to an MLH1 germline mutation presenting with a isocitrate dehydrogenase (IDH) wild-type GB as per the 2021, 5th edition of the World Health Organization (WHO) classification of CNS tumors (it had originally been classified as a grade II astrocytoma as per the 2007, 4th edition of the WHO classification of CNS tumors).24,25 The patient received five lines of therapy before being treated with the anti-PD-1 nivolumab, showing a slow radiological progression. To describe the changes in T-cell presence and clonality induced by treatment, T-cell immunohistochemistry (IHC) and T-cell receptor (TCR) repertoire were analyzed. When comparing the biopsy from initial diagnosis and that performed after progression with nivolumab, no gross changes in the presence of T cells occurred, but a change in TCR clonality was observed. To our knowledge, this is the fifth case reported in the literature of an LS-associated glioma treated with ICIs.
Studies with ICIs in patients with sporadic HGG.

Adj, adjuvant; AEs, adverse events; Bev, bevacizumab; Bev-N, bevacizumab-naïve; Bev-R, bevacizumab-resistant; GBM, glioblastoma; GCs, glucocorticoids; GEP, T-cell-inflamed gene expression profile; HFSRT, hypofractionated stereotactic radiotherapy; HGG, high-grade glioma; HR, hazard ratio; IO, immunotherapy; Ipi, ipilimumab; IRG, immune-regulatory genes; irTRAEs, immune-related TRAEs; MGMT, O6-Methylguanine-DNA Methyltransferase; MGMTmet, MGMT methylated; MGMTunmet, MGMT unmethylated; MSI-H, microsatellite instability-high; mut, mutant; N, number of patients; Neoadj, neoadjuvant; Nivo, nivolumab; ORR, objective response rate; OS, overall survival; PB, peripheral blood; PD, progressive disease; Pembro, pembrolizumab; pGBM, primary GBM; PFS, progression-free survival; rGBM, recurrent glioblastoma; rHGG, recurrent HGG; RS, retrospective series; SD, stable disease; TCR, T-cell receptor; TILs, tumor-infiltrating lymphocytes; TMB, tumor mutational burden; TRAEs, treatment-related adverse events; wt, wild-type; (–), not available.
Glioma cases treated with ICIs and harboring germline or somatic mutations in replication-repair genes.

AA, anaplastic astrocytoma; AOD, anaplastic oligodendroglioma; AWD, alive with disease; AWOD, alive without disease; Bev, bevacizumab; bMMRD, biallelic mismatch-repair deficiency syndrome; CR, complete response; CRC, colorectal cancer; Diax, diagnosis; DWD, dead with disease; FTM, fotemustine; GBM, glioblastoma multiforme; gMMRd, germline MMRd; HmGBM, hypermutated GBM; HGD, high-grade dysplasia; ICI, immune checkpoint inhibitors; IDH, isocitrate dehydrogenase; IDHmut, IDH mutant; IDHwt, IDH wild-type; IHC, immunohistochemistry; IO, immunotherapy; LS, Lynch syndrome; MMRd, mismatch-repair deficient; MSI-H, microsatellite instability-high; Mut, mutant; gMMRd, germline MMRd; N, number of patients; NAL, neoantigen load; Nivo, nivolumab; OD, oligodendroglioma; OS, overall survival; PsPD, pseudoprogressive disease; PD, progressive disease; Pembro, pembrolizumab; PFS, progression-free survival; PR, partial response; rGliomas; recurrent gliomas; RT, radiotherapy; SD, stable disease; sMMRd, somatic MMRd; SX, surgery; TMB, tumor mutational burden; TMZ, temozolomide; W&S, wait and see; (–), not available.
Materials and methods
Ethical considerations
This study was approved by the Institutional Review Board of Hospital Clínico Universitario San Carlos, in accordance with the principles outlined in the ‘World Medical Association Declaration of Helsinki’. A signed informed consent form was obtained from the patient prior to study participation, allowing for publication of this case report, including any accompanying data and images.
Sample collection and processing
The current study used tumor samples from 2013 and 2019. In 2013, a partial tumor resection was undertaken. In 2019, a neuronavigation-assisted tumor biopsy was undertaken. Both tumor samples were formalin-fixed and paraffin-embedded (FFPE).
Immunohistochemistry
IDH1-R132H antibody (clone H09; dilution 1:20), and anti-CD4 (clone 4B12; dilution 1:40), anti-CD8 (clone C8/144B; dilution 1:50), anti-PD-L1 (clone 22C3; dilution 1:50), MLH1 (clone ES05; dilution 1:100), MSH2 (clone FE11; dilution 1:150), MSH6 (clone EP49; dilution 1:50), and PMS2 (clone EP51; dilution 1:50) antibodies (all from Dako North America, Carpintería, CA, USA) were used for studying IDH1 status and T-cell and MMR protein expression by IHC. Positive PD-L1 expression in tumor cells was defined as a membranous staining in at least 1% of tumor cells. 11
DNA extraction
Tumor DNA was obtained from 4 to 8 sections of paraffin-embedded tumor tissue (tumor region was selected by a pathologist) using the ‘QIAamp DNA FFPE Kit GenRead’ kit (QIAGEN, Germantown, MD, USA). This kit removes the cytosine deamination artifacts, minimizing the risk of false single nucleotide polymorphism (SNP) calls. Tumor DNA was quantified using a QUBIT 3.0 fluorometer instrument (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Next-generation sequencing study of IDH1 and IDH2 mutations in tumor tissue
Next-generation sequencing (NGS)-based panel was designed using DesignStudio tool (Illumina, Inc., USA) to optimize target region sequencing coverage. The whole exonic region of IDH1 and IDH2 was sequenced, following the manufacturer’s instructions as previously described. 26 DNA from FFPE tumor samples was quantified using Qubit dsDNA HS Assay (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and 50–150 ng was used for mutational analysis by AmpliSeq methodology (Illumina, Inc., USA). AmpliSeq Library PLUS was used for library preparation (Illumina, Inc., USA), according to the manufacturer’s protocol. After amplification of target regions, primer dimers and partially digest amplicons were digested, and unique combination of index adapters was ligated (AmpliSeq CD Indexes). A second amplification of libraries was performed. Quantity and quality of libraries were checked by Agilent High Sensitivity DNA Kit in an Agilent 2100 Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA, USA). Libraries were diluted to the final loading concentration (7.5–8 pM) and denatured with NaOH for bridge clonal amplification and paired-end sequencing using MiSeq Reagent kit v2 (300 cycles) in a MiSeq instrument (Illumina, Inc., USA). Illumina MiSeq device version 3.1.0.13 (Illumina, Inc., USA) provides alignment (BAM) and variant calling files (vcf). The VariantStudio software (Illumina, Inc., USA) was used to annotate vcf, identifying and classifying the detected genetic variants.
TCR repertoire analysis
The Oncomine TCR-Beta-SR assay (Thermo Fisher Scientific, Waltham, Massachusetts, USA) was used for performing high-throughput NGS to analyze the nucleotide sequence of the CDR3 region coding for the T-cell receptor beta (TCRβ) chain, made up of variable (V), diversity (D), joining (J) (VDJ) and constant regions, following the manufacturer’s instructions. 27 In brief, after isolating and quantifying the DNA, amplicons were obtained after amplification from 100 ng of DNA, then were partially digested and ligated with barcode adapters (Ion Torrent Dual Barcode Kit, Thermo Fisher Scientific, Waltham, Massachusetts, USA) to generate barcoded libraries. Amplification of the purified libraries was performed (five cycles of amplification). Libraries were quantified using Ion Library TaqMan Quantitation Kit and diluted to 25 pM concentration. Libraries were combined to be loaded on an Ion 540 chip using the Ion Chef Instrument and sequenced in the Ion GeneStudio S5 System (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Data analysis was performed using Ion Reporter Software. After removing low-quality, off-target, and error-containing reads from the analysis, VDJ rearrangements were reported, followed by a secondary analysis of the TCRβ repertoire.
MLH1 germline testing
Germline DNA was obtained from peripheral blood leukocytes, and Sanger sequencing was performed in a Applied Biosystems ABI 3130 automated sequencer (Applied Biosystems, Foster City, CA, USA) to confirm the hMLH1 mutation NM_000249:c.1865 T>A; p.(L622H) previously identified in the index family case. Carrier status of our patient was confirmed in peripheral blood from two different blood draws obtained 1 week apart from each other.
Case report
A 34-year-old man, smoker of 40 cigarettes per day, and with grade II obesity, presented to the Emergency Room in August 2012 with an absence of epileptic crisis followed by conjugate gaze deviation and orofacial automatisms, being started on levetiracetam 500 mg/q2wk, with no new episodes. Computed tomography (CT) scan showed a left temporal lobe mass suggesting a low-grade glioma. Brain magnetic resonance imaging (MRI) in September 2012 showed an infiltrating nonenhancing, T1-isointense, T2-hyperintense, and normally perfused (VSCr) left temporal lobe mass invading the cortex and white matter of the parahippocampal convolution, amygdala, and the fusiform and inferior temporal gyruses, compatible with a low-grade diffuse astrocytoma. The tumor mass caused a slight mass effect over the left mesencephalon and minor obliteration of the ipsilateral cisterna ambiens. A repeated brain MRI performed 3 months later showed no apparent changes. In January 2013, a neuronavigation-assisted tumor biopsy was performed confirming the diagnosis of an IDH1-R132H wild-type, low-grade astrocytoma of the left temporal lobe. Sequencing of IDH1 and IDH2 mutations by NGS confirmed the IDH wild-type nature of the tumor. Thereby, it would correspond to an IDH wild-type GB as per the current edition (5th Ed.; 2021) of the WHO Classification of CNS Tumors. The patient informed that his 65-year-old paternal uncle diagnosed with a CRC when he was at the age of 35 years, had been diagnosed of LS after a germline test revealed an hMLH1 pathogenic mutation NM_000249: c.1865 T>A; p.(L622H). The patient’s father, a second paternal uncle and his paternal grandfather, had died from CRC at ages 41, 28, and 50 years, respectively. No other cases of gliomas had been reported in the patient’s family. The patient’s older brother had undergone a right hemicolectomy due to a 4-cm polyp with high-grade dysplasia at the age of 42 years. He had two 35- and 39-year-old male brothers and two 34-year-old twin sisters, and three small children all of them without any history of cancer. The patient underwent germline genetic testing that confirmed the carrier status of the pathogenic hMLH1 mutation previously detected in his paternal uncle (Figure 1). Treatment with temozolomide 150–200 mg/m2/days 1–5 in 28-day cycles was administered for 12 months with good tolerance, showing stable disease as best response. In January 2014, a partial resection was performed due to tumor progression, with pathology informing of a grade III IDH wild-type astrocytoma as per 4th edition of the WHO classification of CNS tumors from 2007, corresponding to an IDH wild-type GB as per the current 5th edition.24,25 Subsequently, the patient received IMRT-based radiotherapy over the tumor mass and safety margins for a total of 60 Gy in 30 fractions (2 Gy per fraction) with good tolerance. Close follow-up ensued until a new brain MRI in February 2015 showed tumor progression. Treatment with fotemustine 80 mg/m2/q2wk was started receiving five cycles but showing a marked tumor progression with clinical deterioration (left hemianopsia, gait instability, and headache) in May 2015. Treatment with bevacizumab 10 mg/kg/q2wk (25 mg/ml solution) was then started 1 month after the last dose of fotemustine. One week after the first cycle, however, the patient was admitted to the Emergency Room due to the absence crisis and aphasia. The patient was diagnosed with a nonconvulsive status epilepticus and transferred to the neurology department where corticosteroids and antiepileptic therapy reverted the episode. Three weeks later, however, treatment with bevacizumab was restarted with no new epileptic episodes and progressive improvement of aphasia and cognitive performance. Brain MRI performed in July 2015, 6 weeks after the first bevacizumab dose, showed a notable radiological improvement, with a complete or near-complete response that kept improving on MRI performed 3 months later, in October 2015. The patient received a total of 27 bevacizumab cycles until February 2017, when, due to the excellent radiological response, treatment was stopped and a ‘wait and see (W&S)’ strategy was started. Close follow-up with MRI every 3 months ensued until April 2018, when tumor progression without clinical deterioration occurred. Treatment with bevacizumab was restarted with slow tumor progression and progressive clinical deterioration (worsening of aphasia and gait instability) after 13 cycles, the last one administered in early November 2018. Due to the absence of standard therapeutic options, and the diagnosis of LS due to a germline hMLH1 mutation NM_000249: c.1865 T > A; p.(L622 H), treatment with the anti-PD-1 agent nivolumab (240 mg/15 days IV; 10 mg/ml solution) via a compassionate use authorization was started, 3 weeks after the last bevacizumab dose. MSI status and TMB analyses were not performed due to the lack of remanent sample. After four cycles, MRI performed in February 2019 showed tumor progression. Because of the doubts regarding a potential tumor pseudoprogression secondary to an immune-mediated effect induced by nivolumab, consent was obtained from the patient to perform a neuronavigation-assisted tumor biopsy, informed as an IDH wild-type GB with a scant immune cell infiltrate, thereby confirming tumor progression. Given the clinical stability and good tolerance, the patient received five additional cycles of nivolumab until May 2019, when an overt clinical and radiological progression occurred. A new line with irinotecan (125 mg/m2/15 days IV) combined with bevacizumab (10 mg/kg/15 days; 25 mg/ml solution) was started 3 weeks after the last dose of nivolumab. Due to toxicity (grade III asthenia, and grade II diarrhea), however, treatment was permanently discontinued after one single cycle administered in June 2019, and the patient was transferred to hospice for end-of-life care where he died 3 months later (Figure 2).
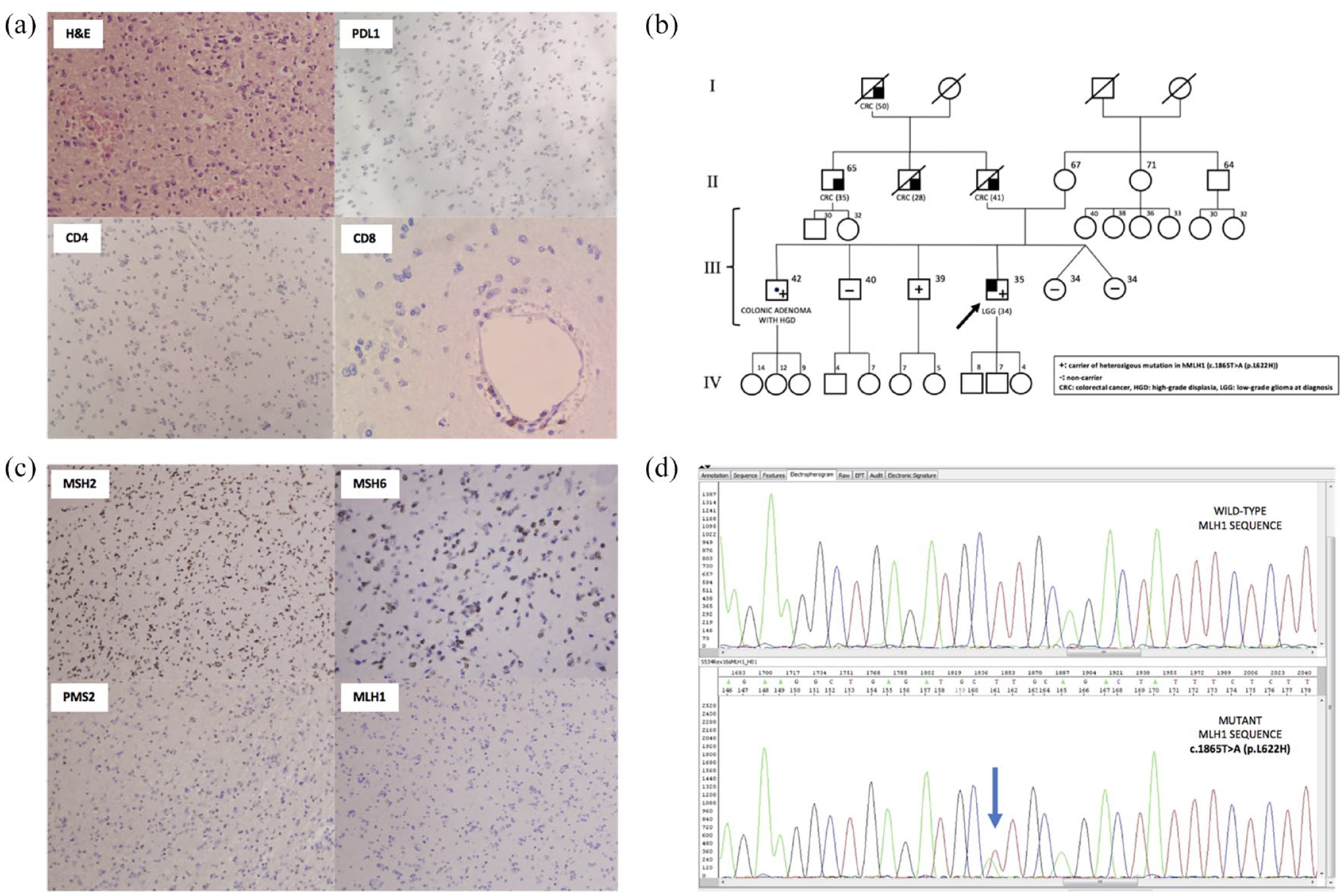
(a) H&E, PD-L1 expression, and T-cell populations in the 2014 tumor biopsy. H&E shows a low-grade glioma, PD-L1 negative, T-CD4 negative and with scant perivascular T CD8+ cells. (b) Pedigree depicting the cancer history of the patient’s family, reflecting an autosomal dominant pattern of inheritance. (c) Mismatch-repair protein IHC shows preserved expression of MSH2 and MSH6, partial PMS2 expression and complete lack of expression of MLH1. (d) Sanger sequence of the patient’s peripheral blood lymphocytes demonstrating a pathogenic MLH1 mutation [c.1865 T > A (p.L622 H)].

MRI tumor evolution from initial diagnosis until the end of therapy.
Results
T-cell and PD-L1 IHC
Biopsies from initial diagnosis in 2013 and after four cycles of nivolumab in 2019 showed a scant immune T-cell infiltration predominantly consisting of perivascular T-CD8+ cells with no apparent changes between the 2013 and 2019 biopsies (Figure 1). Likewise, PD-L1 expression was absent in both the 2013 and 2019 tumor specimens (The 2019 biopsy microphotographs are not shown due to inaccessibility to the slides at the time of writing this article and lack of additional material to repeat the IHC study).
MMR proteins IHC and germline genetic testing
IHC of the tumor biopsy from initial diagnosis showed preserved expression of MSH2 and MSH6, with lack of expression of MLH1 and PMS2 as constituents of the hMutLα complex (Figure 1). 28
Germline testing for hMLH1 mutation NM_000249:c.1865 T > A; p.(L622H) confirmed the carrier status of the patient (Figure 1).
TCR repertoire analysis
The number of TCR clones detected in the 2013 and 2019 biopsies was 26 and 21, respectively. Both Shannon diversity and TCR evenness showed high and comparable values in the 2013 and 2019 biopsies, indicating the good quality of the samples for the analysis. Only one TCR clone was coincident between the two biopsies (V-gene: TRBV12-3, J-gene: TRBJ1-5, CDR3 nucleotide: GCCAGCAGTATTAATTATAGCAATCAGCCCCAGCAT, CDR3 amino acid: ASSINYSNQPQH), although with a higher frequency in the 2019 compared with 2013 specimen (2013 versus 2019 clone frequency: 0.0000006201% versus 0.0000179%) (Table 3, Figure 3, and Supplementary Table).

Comparison of the TCR repertoire between the tumor biopsy from initial diagnosis in 2013 and the tumor biopsy performed in February 2019 after four cycles of nivolumab. (a) TCRB V-gene usage and number of T-cell clones. In the 2013 and 2019 biopsies, 26 and 21 clones were identified, respectively. (b) TCRB V-gene usage and evenness. Evenness was 0.7233 and 0.7049 in 2013 and 2019. (c) TCRB V-gene usage and Shannon diversity, the latter achieving 3.3997 and 3.0962 in 2013 and 2019, respectively. (d) Bar plot depicting the number of clones detected per V-gene. (e) Bar plot showing the number of reads per V-gene allele.
Summary of number of TCR clones, Shannon diversity, and Evenness in the biopsies from initial diagnosis in 2013 and that performed in 2019 after four cycles of nivolumab.

Discussion
Our case is notable for constituting a rare presentation of LS, that is, the development of an IDH wild-type GB in a patient harboring a germline MLH1 mutation. MLH1-driven LS is an autosomal dominant disease characterized by a 40–60% risk of CRC, a 2–12% risk of urinary tract cancer, and, in females, a 30–50% risk of endometrial cancer, and a 5–15% risk of ovarian cancer. Rarely (<2%), patients may develop glial tumors.29,30 LS is associated with MMR defects leading to MSI, and thereby to hypermutant cancers with a high neoantigen load, which make these tumors easily recognizable by the immune system, and specifically by T cells. 31 This explains why LS-associated cancers show a high objective response rate (ORR) when treated with immunotherapy. While ORR to anti-PD-1 agents alone or in combination with anti-CTLA4 agents in metastatic CRC range from 30% to 60%, only a few cases of LS-associated gliomas treated with anti-PD-1 agents have been reported, demonstrating either objective responses or durable disease control (Table 2).31–33 To our knowledge, only four other LS-associated gliomas treated with anti-PD-1 agents have been reported to date. In only one of them, the response to single-agent PD-1-inhibition (pembrolizumab) was properly evaluated, showing stable disease as best response and an overall survival of at least 12 months, while the other three reported cases either combined nivolumab with radiotherapy followed by the maintenance nivolumab or used (neo)adjuvant anti-PD-1 agents prior and/or after rescue surgery for newly diagnosed and recurrent GB. Interestingly, two of the latter three cases were alive without disease 20 and 60 months after starting anti-PD-1 therapy (Table 2).17–19
Similar to the case reported by Kamiya-Matsuoka et al., 17 our patient did not respond to anti-PD-–1 inhibition, but a slowly progressive disease with a 10-month overall survival was achieved, which suggests a true benefit from nivolumab, given the 9.8-month median OS with nivolumab in second-line recurrent unselected GB patients, while our patient received nivolumab as a sixth line of therapy. 11 Therefore, the heavily treated nature of the tumor of our patient may explain the lack of response and limited survival. The initial biopsy from 2013 and the biopsy performed after four cycles of nivolumab in 2019, however, showed almost complete absence of T cells, thereby suggesting an only minor, at best, antitumor immune response.
We studied the TCR repertoire in the original tumor biopsy from 2013 and the post-nivolumab biopsy from 2019 and found a similar number of T-cell clones, but a completely different T-cell repertoire, with only one T-cell clone being concordant between the two biopsies that significantly increased in the 2019 specimen compared with the prior one from diagnosis. This may be explained because of the high spatial and temporal heterogeneity known to occur in GB, which is a consequence of time and treatment pressure. 34 We cannot assume that these changes in TCR clonality occurred in response to nivolumab since we could not perform a tumor biopsy immediately prior to the start of anti-PD-1 therapy to make a direct comparison of pre- and post-nivolumab TCR repertoires. Interestingly, other studies have shown that a certain TCR profile may be prognostic in GB patients receiving a personalized antitumor vaccine. 35 In addition, it would have been of interest to study the tumor mutational burden and MSI in our patient’s tumor, which, at least in theory, should be TMB-high and MSI-H as it commonly occurs in LS-associated cancers.18,19,31
Other heritable syndromes leading to hypermutant cancers that also commonly respond to ICIs are the BMMRDS characterized by the occurrence of gliomas and hematological cancers at early ages, and polymerase deficiency syndrome due to POL-E and POL-D mutations where both colorectal and endometrial cancers but also gliomas are part of the disease spectrum.13,14,36 A few cases of gliomas treated with anti-PD-1 agents developed within the BMMRDS and germline POLE-mutant syndrome have been reported. Interestingly, responses to PD-1-inhibitors in these BMMRDS-associated gliomas were dramatic and durable in all the four reported cases, suggesting that these pediatric tumors may be specially prone to respond to these agents.13,15,16 As reported in two of these BMMRD patients, a rapid tumor flare may occur at the start of treatment that should be closely monitored and treated with steroids and anticonvulsants before resuming immunotherapy. 13 Finally, the sole reported case of germline POLE-mutant GB treated with ICIs showed a paradoxical response and developed inflammatory changes after pembrolizumab therapy. 14
While other immunotherapy modalities, such as CAR-T-cells, vaccines, and oncolytic viral therapy, may be promising in selected patients with gliomas, none of these have been evaluated in gRRD-associated brain tumors, a disease niche where it would be worth evaluating them.37–41
Our patient experienced an almost 3-year progression-free survival (PFS) after starting bevacizumab in third line which is much longer than the 4.2 months PFS reported in unselected patients with recurrent GB. 42 While a component of radionecrosis (RN) was present at the time of starting this therapy, and RN is known to be associated with brilliant responses after only four cycles, in our case, there was also a tumoral component as suggested by the very high perfusion values. Therefore, while the near-complete and durable response to bevacizumab may be partially explained by the response of the RN component, the almost 3-year-long response of the tumor component is notable and leads to hypothesize a potential immune-modulatory effect driven by bevacizumab and linked to the MMRd status of the tumor that should be investigated. 43 Indeed, a synergistic effect from anti-PD-1 agents combined with antiangiogenics has been demonstrated in other entities, such as kidney cancer or hepatocellular carcinoma, where both types of agents are commonly used.44–47 It would have, however, been of interest to study if the tumor belonged to an intrinsic molecular subtype prone to respond to bevacizumab, and other molecular factors related to the angiogenesis process that would explain the response to bevacizumab. 48
Conclusion
gRRD gliomas are exceptional disease entities that, contrary to sporadic gliomas, seem to benefit from anti-PD-1 therapy. Our case is notable for constituting the fifth LS-associated glioma treated with PD-1-inhibitors. While no response was achieved, a slowly progressive disease was observed and a change in TCR clonality occurred under treatment with nivolumab. Although a rare disease entity, gRRD gliomas could serve as a model to better understand the role of the immune system in the development and treatment of CNS tumors.
Supplemental Material
sj-xlsx-1-tam-10.1177_17588359221100863 – Supplemental material for Durable benefit and change in TCR clonality with nivolumab in a Lynch syndrome–associated glioma
Supplemental material, sj-xlsx-1-tam-10.1177_17588359221100863 for Durable benefit and change in TCR clonality with nivolumab in a Lynch syndrome–associated glioma by Santiago Cabezas-Camarero, Rebeca Pérez-Alfayate, Vanesa García-Barberán, María Carmen Polidura, María Natividad Gómez-Ruiz, Isabel Casado-Fariñas, Issa Ahmad Subhi-Issa, José Carlos Plaza Hernández, Pilar Garre, Isabel Díaz-Millán and Pedro Pérez-Segura in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
The authors thank the Grupo Español de Investigación en Neuro-Oncología (GEINO) for funding this study.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the ‘BECA GEINO 2016’ grant, from Grupo Español de Investigación en Neuro-Oncología (GEINO).
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.