
Abstract
Background:
Ovarian carcinoma is extremely sensitive to (platinum-based) chemotherapy; however, most patients will relapse with platinum-resistant disease, badly affecting their prognosis. Effective therapies for relapsing resistant tumors are urgently needed.
Methods:
We used patient-derived xenografts (PDXs) of ovarian carcinoma resistant to cisplatin (DDP) to test in vivo the combination of paclitaxel (15 mg/kg i.v. once a week for 3 weeks) and onvansertib, a plk1 inhibitor, (50 mg/kg orally 4 days a week for 3 weeks). The PDX models were subcutaneously (s.c.) or orthotopically transplanted in nude mice and antitumor efficacy was evaluated as tumor growth inhibition and survival advantages of the combination over untreated and single agent treatment.
Results:
The combination of onvansertib and paclitaxel was very well tolerated with weight loss no greater than 15% in the combination group compared with the control group. In the orthotopically transplanted PDXs, single onvansertib and paclitaxel treatments prolonged survival; however, the combined treatment was much more active, with median survival from three- to six-fold times that of untreated mice. Findings were similar with the s.c. transplanted PDX, though there was greater heterogeneity in tumor response. Ex vivo tumors treated with the combination showed greater induction of γH2AX, marker of apoptosis and DNA damage, and pSer10H3, a marker of mitotic block.
Conclusion:
The efficacy of onvansertib and paclitaxel combination in these preclinical ovarian cancer models supports the clinical translatability of this combination as an effective therapeutic approach for platinum-resistant high-grade ovarian carcinoma.
Introduction
Epithelial ovarian cancer (EOC) is the most lethal gynecological cancer. 1 The most common EOC is the high-grade serous (HGS) histotype, accounting for more than 70% of the diagnosed tumors. 2 HGS is very sensitive to chemotherapy, 3 probably because more than 50% of these tumors have defects in homologous recombination repair,4,5 involved in the repair of platinum-induced DNA lesions. 6 However, beside the high rate of response to a platinum/paclitaxel first-line adjuvant therapy, 70–80% of patients will relapse with a much less platinum-responsive tumor. 3 Platinum resistance will develop and this is still one of the major obstacles in the treatment of relapsed ovarian cancer. 6
For many years, the principal prognostic factor in recurrent ovarian carcinoma and the main parameter for guiding therapeutic decisions has been the platinum-free interval (PFI), clustering patients as platinum-refractory (with disease recurrence during chemotherapy), platinum-resistant (progressing within 6 months from the end of first-line platinum-based therapy), potentially platinum-sensitive (recurrence between 6 and 12 months), and platinum-sensitive (experience recurrence after 12 months).7,8
However, this definition was recently challenged in the 2019 European Society for Medical Oncology–European Society of Gynecological Oncology (ESMO–ESGO) guidelines and partially replaced with the concept of the treatment-free interval, with patients conceptually categorized in those eligible for a platinum re-challenge and those for whom platinum is not an option.9,10 In patients for whom platinum is no longer an option, the therapeutic approach is single chemotherapy, with agents that have shown activity.10,11 Second-line treatments include topotecan, gemcitabine, pegylated liposomal doxorubicin (PDL), and paclitaxel. 12 Recently, the addition of bevacizumab to second-line therapy improved the patients’ reported outcomes and their quality of life, 13 leading to approval of the combination of bevacizumab and non-platinum chemotherapy in Europe and the United States for patients not previously treated with bevacizumab. 10
Polo-like kinase 1 (Plk1) is a serine–threonine protein kinase with a key role in regulating mitotic events.14,15 Evidence is accumulating on its role in non-mitotic events as well, 16 such as in DNA damage responses by regulating different sensor proteins and DNA repair pathways.17–20 Plk1 is overexpressed in human tumors and correlates with poor patient prognosis. 21 Plk1 inhibitors are now under clinical testing.22–24 Onvansertib is an orally available, highly selective Plk1 inhibitor under clinical investigation. 25 It has been reported to be active in combination with paclitaxel in different preclinical models, with very interesting synergistic activity.26,27
Considering that paclitaxel is indicated as second-line therapy in platinum-resistant ovarian relapses, we tested the combination of paclitaxel and onvansertib in patient-derived xenografts (PDX) resistant to cisplatin (DDP). In most of the models used, antitumor activity was better with the combination than with the single drugs, supporting the use of this combination in platinum-resistant ovarian carcinomas.
Materials and methods
Animals and drugs
Five-week-old female NCr-nu/nu mice were obtained from Envigo Laboratories (Italy) and maintained under specific pathogen-free conditions, housed in isolated vented cages, and handled using aseptic procedures. Procedures involving animals were conducted in conformity with the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D. lg 26/2014; authorization no.19/2008-A issued 6 March 2008 by the Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System Certificate: UNI EN ISO 9001:2015, reg. no. 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition); and EU directive and guidelines (EEC Council Directive 2010/63/UE). An institutional review board and the Italian Ministry of Health approved all the in vivo experiments performed with PDXs. (approval no. 475/2017-PR). Tumor fragments were subcutaneously (s.c.) transplanted (MNHOC124) or tumor cell suspensions (approximately 15 × 106cells/mouse for MNHOC266R and 10 × 106cells/mouse for MNHOC76) were orthotopically implanted (intraperitoneally, i.p.). Animals were randomized (eight–ten/group) when tumors reached approximately 100–150 mg in s.c transplanted PDXs or 10 days after i.p. tumor cells injection in different experimental groups (control, onvansertib, paclitaxel, or combination). Onvansertib was given orally at the dose of 50 mg/kg for four consecutive days for three cycles with 3 days of rest; paclitaxel was injected intravenously (i.v.), 15 mg/kg once a week for 3 weeks. For the combined treatment, paclitaxel was injected 2 h after the last onvansertib treatment dose.
Tumor growth was measured twice weekly with a Vernier caliper, and tumor weights (mg = mm3) were calculated as follows: (length (mm) × width2 (mm2))/2 where width < length); body weights were recorded and considered an indirect parameter of drug toxicity. For i.p, transplanted tumors, mice were weighted three times a week and the appearance of ascites was recorded.
The efficacy of treatment was evaluated for s.c. PDXs as the best tumor growth inhibition (%T/C = (mean tumor weight of treated tumors/mean tumor weight of control tumors) × 100) and for i.p. transplanted PDX as increase of lifespan (ILS% = ((median survival days of treated mice–median survival days of control mice)/median survival days of treated mice) × 100. T/C value < 42% and ILS > 25% were considered the minimum for antitumor activity, in accordance with published criteria. 28
For pharmacodynamics studies, tumor bearing mice were treated with the single or combined drugs following the schedules used in the experiment. However, mice were given just one cycle then euthanized at 2 and 24 h later the last dose of onvansertib for the single group and 24 h after paclitaxel in both the single-treatment and combination groups. The tumors were removed and snap-frozen. The frozen samples were homogenized in protein lysis buffer, loaded on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotted.
Onvansertib was dissolved in vehicle (0.5% methylcellulose Tween-20 1%) freshly on days of dosing. Paclitaxel (Indena S.p.a., Milan, Italy) was dissolved in 50% Cremophor EL (Sigma-Aldrich) and 50% ethanol and further diluted with saline before use. 28
Western blot analysis
Snap tumor fragments were lysed in ice-cold whole cell extract buffer containing 50 mM TrisHCl pH 7.4, 250 mM NaCl, 0.1% Nonidet NP40, 5 mM EDTA and NaF 50 mM with a protease inhibitor cocktail (Sigma). Lysates were cleared by centrifuging at 12,000 rpm for 5 min. Cell lysates containing equal amount of protein (30–70 µg) were resolved on 10–12% SDS-PAGE gels. The proteins were then transferred to nitrocellulose membranes (PROTRAN, Schleicher and Shull).
Immunoblotting was carried out with the following antibodies and visualized using Odyssey FC Imaging System (Li-COR): anti-actin (C-11) #sc1615 provided by Santa Cruz Biotechnology. Anti-phospho-Histone H3 (Ser10) (6G3) #9706 was purchased from Cell Signaling Technology. Anti H2AX pSer139 (#05-636) was purchased from Millipore. The secondary antibody anti-goat (sc-2354) was purchased from Santa Cruz Biotechnology (Heidelberg, Germany). The anti-rabbit (#1706515) and anti-mouse (#1706516) were conjugated with horseradish peroxidase (HRP), were purchased from Bio-Rad Laboratories S.r.l.
Caspase-3 activity assay
Caspase-3 activity was measured by an enzymatic assay using a fluorogenic substrate for caspase-3, Ac-DEVD-AMC (acetyl Asp–Glu–Val–Asp 7–amido-4-methylcoumarin (AMC). Protein extracts were mixed with the apoptosis buffer (Hepes pH 7.5 20 mM, glycerol 10%, and dithiothreitol (DTT) 10 mM) in a white 96-well plate and incubated at 37°C for 5 min. The substrate was then added at a final concentration of 12.5 µM. Fluorescent AMC production was measured at excitation 370 nm, emission 460 nm, using a plate reader (Infinite M200, TECAN). The activity of each sample was examined in duplicate. Activity was expressed as the linear change in fluorescence units per hour and normalized for the protein concentration.
Statistical analysis
Statistical significance was determined with GraphPad Prism 7.05 (GraphPad Software). Figures legends specify which test was used.
Results
PDX models
Table 1 summarizes the characteristics of the models used: MNHOC266R (cisplatin, DDP-resistant), MNHOC76 (DDP-resistant) and MNHOC124R (DDP-resistant), from here denominated #266R, #76, and #124R. All the PDXs are high-grade serous ovarian carcinomas (HGSOCs). We selected one PDX model with primary DDP resistance (#76) and two with acquired resistance to DDP (#124R and #266R). #124R and #266R were obtained through multiple in vivo DDP rounds (six to eight DDP cycles) of treatment, as reported.29,30 The main histological and molecular characteristics of the PDXs under studied are summarized in Supplementary Table 1.
Characteristics of the PDXs used in this study.

HGSOC, high-grade serous ovarian carcinoma; i.p., intraperitoneally; na, not available; R, resistant; s.c., subcutaneously; wt, wild type.
Onvansertib and paclitaxel combined in orthotopically transplanted PDXs
Figure 1 shows the Kaplan Mayer curves of #266R (panel a) and #76 (panel b) transplanted mice treated with onvansertib or paclitaxel as single agents and their combination. Paclitaxel/onvansertib combination was safe and well tolerated as demonstrated by the lack of significant body weight loss (Supplementary Figure 1). Single onvansertib and paclitaxel prolonged survival (Figure 1 and Table 2), but the combined treatment was much more active. In fact, in both #266R and #76 models, mice treated with the combination survived longer, with a median survival of 82 and 304 days, respectively (Table 2), or approximately three and six times the control median survival (24.5 and 49 days). Of note, the number of long-term survivors without tumors was higher in the combination treated mice in both models (one mouse in the combination #266R treated group and four out of eight mice in the #76 treated mice) versus none in the other #266 experimental groups and only one mouse in single onvansertib and placlitaxel treated #76 bearing mice. The combination group survival curves show a significant advantage over controls and over both single agents (Figure 1).
Antitumor efficacy of the treatments in #266R, #76, and #124R xenografts.


Survival curve of mice transplanted with (a) #266R and (b) #76 in the different experimental groups. (a) Log-rank (Mantel–Cox) in #266R. Mice transplanted intraperitoneally with #266R xenograft were randomized to receive vehicle (ctrl -•-), onvansertib (-■-), paclitaxel (-▲-), or their combination (-▼-). Ctrl versus onvansertib, ***p < 0.001; ctrl versus paclitaxel, **p < 0.01; ctrl versus combination, ****p < 0.0001; paclitaxel versus combination, *p < 0.05; onvansertib versus combination, ***p < 0.001. (b) Log-rank (Mantel–Cox) in #76. Mice transplanted intraperitoneally with #76 xenograft were randomized to receive vehicle (ctrl -•-), onvansertib (-■-), paclitaxel (-▲-), or their combination (-▼-). Ctrl versus onvansertib, ** p < 0.0017; ctrl versus paclitaxel, ****p < 0.0001; ctrl versus combination, ****p < 0.0001; combination versus paclitaxel, *p < 0.037; combination versus onvansertib, **p < 0.0098.
Onvansertib and paclitaxel combination in an s.c. transplanted PDX
We then tested the combination in s.c. implanted #124R ovarian carcinoma PDX model with acquired resistance to DDP. 29 Again, the combination was well tolerated with weight loss no greater than 15% in the combination group compared with the control group (Supplementary Figure 2). Onvansertib treatment only marginally inhibited tumor growth (Figure 2(a)) with no increase in mean survival time in #124R bearing mice (Figure 2(b) and Table 2). Paclitaxel inhibited tumor growth (Figure 2(a)) with significant increment in survival of mice bearing #124R PDX (Figure 2(b) and Table 2). When the two drugs were combined, the antitumor effect was greater even though not significantly different from the single paclitaxel treatment. Indeed, in this latter experimental group, tumor responses were heterogeneous: marked tumor stabilization was seen in two of the six randomized animals, that lasted 30 days after the last drug treatment; tumor regrew in just one mouse, much more slowly than in the control, while no tumor was found in the remaining mouse. Both mice were euthanized on day 191 post-transplantation (Supplementary Figure 3). These data suggest a longer lasting effect of the combination on tumor growth than paclitaxel single agent.

Antitumor activity of onvansertib, paclitaxel and their combination in the #124R model. (a) #124R xenografts were transplanted subcutaneously and when tumor masses reached 100–150 mg, mice were randomized to receive vehicle (ctrl -•-), onvansertib (-■-), paclitaxel (-▲-), or their combination (-▼-). Data are the mean ± SE of tumor masses, as described in the “Materials and Methods” section; each group consisted of 8–10 animals. (b) Log-rank (Mantel–Cox) in #124R. (ctrl -•-), onvansertib (-■-), paclitaxel (-▲-), or their combination (-▼-). Ctrl versus onvansertib, *p < 0.0029; ctrl versus paclitaxel, **p < 0.0025; ctrl versus combination, **p < 0.001; combination versus onvansertib, *p < 0.024.
Combined paclitaxel/onvansertib induces mitotic arrest and apoptosis
To investigate the mechanisms underlying the combinatorial effects of paclitaxel and onvansertib, we analyzed tumors of mice treated or not with drugs as single agents or the combination at the doses used for antitumor activity. Mice were euthanized 2 and 24 h after onvansertib, and 24 h after paclitaxel given alone or after a 3-day pre-treatment with onvansertib. We looked for induction of pSer10 H3 and γH2AX, respectively, markers of a mitotic block 31 and DNA damage, including apoptotic DNA fragmentation due to apoptosis.32,33
In the #226R orthotopically transplanted PDX, pSer10 H3 was clearly induced only in the combination group at 24 h, while γH2AX seemed higher at 24 h in the combination-treated group than in single-agent-treated group, even if a statistical difference could not be observed probably due to a high inter-tumor sample variability (Figure 3(a), left panel; Supplementary Figure 4). Again, a greater apoptosis induction, as detected by the increase in caspase-3 activation, was found in the combination-treated group (Figure 3(a), right panel). These data seem to parallel the greater antitumor activity in the mice treated with the combination.
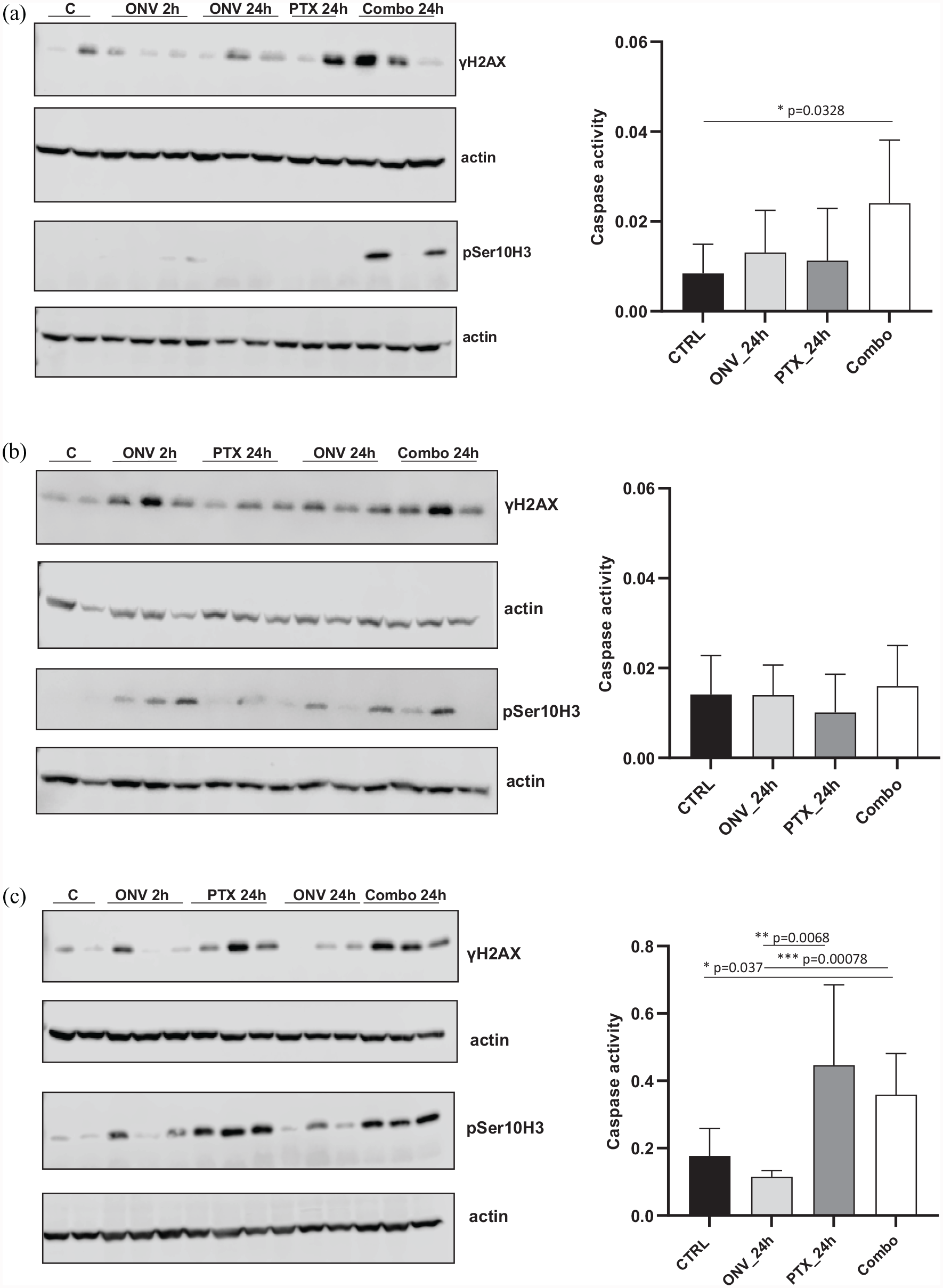
In vivo pharmacodynamic assessment of mitotic block, DNA damage and apoptosis in PDXs treated or not with the single or combined drugs. Western blot analysis showing pSer10 H3 and γH2AX protein levels in xenograft tumor protein extracts in #266R (a, left panel), in #76 (b, left panel), and #124R (c, left panel). Two or three replicates for each condition were used. Caspase-3 activity in tumor tissue extracts from mice of different PDX models, treated or not, was evaluated as described in the “Materials and Methods” section, is shown in the corresponding right panels (a: #266R; b: #76; and c: #124R) Data are the mean
Less clear was the preferential induction of pSer10 H3 and γH2AX in both #76 and #124 PDX models in the combination-treated tumor samples at the 24 h (Figure 3(b) and (c), left panels), probably due to the fact that biomarkers induction was also seen after onvansertib and paclitaxel single-agent-treated tumor samples. No apoptosis induction could be observed in the #76 PDX model, while in the #124 model, a clear induction could be observed in the paclitaxel and combination-treated tumor samples (Figure 3(b) and (c), and Supplementary Figure 4). This might be because paclitaxel as single agent was much more active in this model than in #266R and #76. Two hours after onvansertib treatment, in two out of three models, induction of pSer10H3 could be clearly observed, suggesting that at this dose, onvansertib could induce a G2-M block and possibly sensitize cells to paclitaxel treatment.
Discussion
This study investigated the therapeutic effects of the paclitaxel and onvansertib combination in ovarian carcinoma PDXs resistant to DDP. Platinum-based therapy is still the gold standard in HGSOC. 3 However, despite the high rate of tumor remissions, most patients will develop a tumor recurrence resistant to platinum, that is, increasingly difficult to treat. In platinum-resistant relapses, the sequential use of non-platinum compounds is considered the standard.8,10 The most frequently used drugs are PDL, paclitaxel, gemcitabine, and topotecan, reported to give similar response rates, progression-free survival and overall survival. 8 The choice of the agent depends on many factors (prior therapies, residual toxicities, patient’s preference, etc.). Recently, a weekly paclitaxel regimen has given response rates between 20% and 30%, with less neurotoxicity than standard schedule (every 3 weeks), and has increasingly becoming the control arm in randomized trials of refractory/resistant ovarian cancers.34,35 Combinations of paclitaxel and saracatinib (an Src inhibitor) and linsitinib (a dual insulin receptor and insulin-like growth factor-1 receptor inhibitor) have shown promising preclinical activity; however, the clinical result in platinum-resistant tumors was no better than paclitaxel alone.36,37 Recently, a phase I trial of rapamycin (mTOR inhibitor) combined with paclitaxel reported a 52% response rate in platinum-resistant HGSOCs and this combination is now being explored in the OCTOPUS trial, 38 whose results are awaited.
We focused on the combination of paclitaxel and onvansertib in platinum-resistant ovarian carcinoma using validated preclinical models, for four main reasons. The first was that in this setting there is an urgent need for new active therapies and paclitaxel is a recognized active drug to be potentially combined with other agents; the second reason is that the specific paclitaxel and Plk1 inhibitor combination has been reported active in different preclinical models of Ewing sarcoma, rhabdomyosarcoma, and triple-negative breast cancer models.27,39,40 We also recently reported that the paclitaxel and onvansertib combination was synergic in both in vitro and in vivo models of mucinous ovarian carcinoma. 26 The antitumor activity of the paclitaxel and onvansertib combination can be further conceptualized on the basis of recent data suggesting that platinum-resistant ovarian cells are more susceptible to mitotic catastrophe induced by inhibiting the mitotic regulator Plk1 and that these resistant cells are functionally dependent on Plk1 kinase activity. 41 Finally, the non-overlapping dose-limiting toxicities of the two drugs (neutropenia and thrombocytopenia for onvansertib and neurotoxicity for paclitaxel25,42,43) present an advantage for this combination in clinical settings.
Using ovarian PDX models available in our laboratory,28,29,44 we selected models with primary and acquired resistance to platinum-based therapy. All the PDXs were TP53 mutated and this could be important as some studies have suggested that TP53 mutated cells are more responsive to a Plk1 inhibitor and paclitaxel than wild-type TP53 cells.45,46 Onvansertib is a new oral inhibitor of Plk1 undergoing clinical investigation in solid tumors and leukemia.25,43 In the orthotopically transplanted tumor models, paclitaxel/onvansertib combination gave a clear survival advantage, as mice treated with both drugs survived longer than after single agent treatment. In the #124R model, while the tumor responses to the combination of onvansertib and paclitaxel were heterogeneous, two out of six mice showed a lasting tumor response and, in one case, the tumor regressed completely and the mouse was sacrificed without tumor. As a whole, these data suggest that this combination warrants further preclinical investigation and possibly clinical confirmation.
We addressed the molecular mechanism of the greater antitumor effect of the combined treatment. Considering that both drugs are able to induce a mitotic block, we looked for pSer10 H3 induction and found that it was clearly more induced as compared with single drugs in #266R model, while less clear was the pSer10 H3 preferential induction in the other two models. Similar considerations can be done for the induction of apoptosis (detected both as γH2AX and caspase-3 activation) that tended to be higher at 24 h in the dual-treated mice bearing #266R than in single-drug-treated mice. Cells arrested in mitosis by paclitaxel or Plk1 inhibitors can undergo apoptosis during mitosis or mitotic slippage, after which cells can arrest, cycle or eventually die26,47–49 and our data suggest that this could be occurring in our experimental conditions.
In a phase II trial, volasertib, a potent, pan-Plk inhibitor, 50 had reported activity in heavily pre-treated patients with relapsed ovarian cancer refractory or resistant to platinum-based therapies. 51 The side effects of Plk1 inhibitors are mainly hematological and seem not to overlap paclitaxel neurotoxicity, further supporting the advantage of this combination.
Our data uphold the clinical translatability of the onvansertib and paclitaxel combination as an effective therapeutic approach in platinum-resistant HGSOC.
Supplemental Material
sj-docx-1-tam-10.1177_17588359221095064 – Supplemental material for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas
Supplemental material, sj-docx-1-tam-10.1177_17588359221095064 for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas by Roberta Affatato, Michela Chiappa, Federica Guffanti, Francesca Ricci, Laura Formenti, Robert Fruscio, Marta Jaconi, Maya Ridinger, Mark Erlander and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359221095064 – Supplemental material for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas
Supplemental material, sj-docx-2-tam-10.1177_17588359221095064 for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas by Roberta Affatato, Michela Chiappa, Federica Guffanti, Francesca Ricci, Laura Formenti, Robert Fruscio, Marta Jaconi, Maya Ridinger, Mark Erlander and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-pptx-1-tam-10.1177_17588359221095064 – Supplemental material for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas
Supplemental material, sj-pptx-1-tam-10.1177_17588359221095064 for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas by Roberta Affatato, Michela Chiappa, Federica Guffanti, Francesca Ricci, Laura Formenti, Robert Fruscio, Marta Jaconi, Maya Ridinger, Mark Erlander and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-pptx-3-tam-10.1177_17588359221095064 – Supplemental material for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas
Supplemental material, sj-pptx-3-tam-10.1177_17588359221095064 for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas by Roberta Affatato, Michela Chiappa, Federica Guffanti, Francesca Ricci, Laura Formenti, Robert Fruscio, Marta Jaconi, Maya Ridinger, Mark Erlander and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-pptx-4-tam-10.1177_17588359221095064 – Supplemental material for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas
Supplemental material, sj-pptx-4-tam-10.1177_17588359221095064 for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas by Roberta Affatato, Michela Chiappa, Federica Guffanti, Francesca Ricci, Laura Formenti, Robert Fruscio, Marta Jaconi, Maya Ridinger, Mark Erlander and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-pptx-5-tam-10.1177_17588359221095064 – Supplemental material for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas
Supplemental material, sj-pptx-5-tam-10.1177_17588359221095064 for Onvansertib and paclitaxel combined in platinum-resistant ovarian carcinomas by Roberta Affatato, Michela Chiappa, Federica Guffanti, Francesca Ricci, Laura Formenti, Robert Fruscio, Marta Jaconi, Maya Ridinger, Mark Erlander and Giovanna Damia in Therapeutic Advances in Medical Oncology
Footnotes
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The generous contributions of AIRC (The Italian Association for Cancer Research) are gratefully acknowledged (IG 19797 to GD). They are grateful to Cardiff Oncology for financial support and for providing onvansertib.
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RM and EM are employees and shareholders of Cardiff Oncology. All the other authors have no competing interests.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.