
Abstract
Background:
Epithelial ovarian cancer is the most lethal gynecological cancer and the high mortality is due to the frequent presentation at advanced stage, and to primary or acquired resistance to platinum-based therapy.
Methods:
We developed three new models of ovarian cancer patient-derived xenografts (ovarian PDXs) resistant to cisplatin (cDDP) after multiple in vivo drug treatments. By different and complementary approaches based on integrated metabolomics (both targeted and untargeted mass spectrometry-based techniques), gene expression, and functional assays (Seahorse technology) we analyzed and compared the tumor metabolic profile in each sensitive and their corresponding cDDP-resistant PDXs.
Results:
We found that cDDP-sensitive and -resistant PDXs have a different metabolic asset. In particular, we found, through metabolomic and gene expression approaches, that glycolysis, tricarboxylic acid cycle and urea cycle pathways were deregulated in resistant versus sensitive PDXs. In addition, we observed that oxygen consumption rate and mitochondrial respiration were higher in resistant PDXs than in sensitive PDXs under acute stress conditions. An increased oxidative phosphorylation in cDDP-resistant sublines led us to hypothesize that its interference could be of therapeutic value. Indeed, in vivo treatment of metformin and cDDP was able to partially reverse platinum resistance.
Conclusions:
Our data strongly reinforce the idea that the development of acquired cDDP resistance in ovarian cancer can bring about a rewiring of tumor metabolism, and that this might be exploited therapeutically.
Introduction
Epithelial ovarian cancer (EOC) is the most lethal gynecological cancer with more than 14,000 deaths/year in western countries. 1 The high mortality is mostly due to the frequent presentation at advanced stage, and to primary or acquired resistance to platinum-based therapy. Different studies based on whole-genome, proteomic or transcriptomic profiling studies have defined some of the mechanisms involved in the development of platinum resistance in ovarian cancer.2–4 However, these results have not yet been translated into effective therapeutic strategies to prevent or overcome platinum resistance.
Metabolism has recently emerged as a new potential therapeutic target in oncology, and different trials are currently ongoing targeting altered tumor metabolic pathways. 5 Accumulating evidence suggest not only that tumor metabolism differs from that of matched normal tissues,6,7 but also that metabolic reprogramming may indeed cause therapy resistance.6,8,9 Proteomic analysis has been performed in ovarian cancer cell lines or patient samples (biopsies, plasma specimens) to characterize the metabolic profile associated with cDDP resistance.10,11 Even if expression of some proteins correlated with resistance, a defined metabolic phenotype characterizing cDDP resistance is still lacking. Alterations in the methionine degradation super pathway and cysteine biosynthesis segregated cDDP-resistant from cDDP-sensitive ovarian cells 12 and low serum phospholipids and essential amino acids were correlated with a worse outcome in ovarian patients. 13 Again, specific metabolic signatures were associated with chemoresistance,14–17 but unfortunately these results are often contradictory and a long way from a clinical application.
We report here the metabolic profile of cDDP-resistant ovarian cancer patient-derived xenografts (PDXs), obtained from cDDP-sensitive PDXs by in vivo repeated drug treatment. As most of the data on metabolism reprograming and resistance to therapy have been generated in in vitro systems, our in vivo models are more likely to be the clinically relevant setting to investigate the role of metabolism in platinum resistance. We used different and complementary approaches based on integrated metabolomics (targeted and untargeted), gene expression, and functional assays to explore the metabolic scenario associated with in vivo acquired cDDP resistance. The data generated led to a therapeutic intervention based on the combination of metformin and cDDP that was able to reverse cDDP resistance in vivo.
Methods
In vivo studies
Animals
Female NCr-nu/nu mice obtained from Envigo Laboratories (Udine, Italy) were used when they were 6- to 8-weeks old. Mice were maintained under specific pathogen-free conditions, housed in isolated vented cages, and handled using aseptic procedures. The Istituto di Ricerche Farmacologiche Mario Negri IRCCS, adheres to the principles set out in the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D. lg 26/2014; authorization no.19/2008-A issued 6 March 2008 by the Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System Certificate: UNI EN ISO 9001:2008, reg. no. 6121); the National Institute of Health (NIH) Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directive and guidelines (European Economic Community [EEC] Council Directive 2010/63/UE). An institutional review board and the Italian Ministry of Health approved all the in vivo experiments performed with PDXs (authorization no. 705/2016-PR).
Isolation of cDDP-R ovarian cancer PDXs
Three high-grade serous/endometrioid cDDP sensitive(s) PDXs were selected and made cDDP resistant. Specifically, tumors were subcutaneously transplanted into nude mice, and when they reached a tumor weight of ~150 mg, mice were treated with multiple cycles of cDDP (each cycle consisting in cDDP given intravenously (i.v.) weekly for 3 weeks (q7x3) at the dose of 5 mg/kg). When tumor weights reached the ethical limits (10% of mice body weight), mice were sacrificed, and tumors transplanted into other mice to receive a new cycle of cDDP. After a total of five to seven cDDP cycles, we obtained three PDX models that were cDDP-resistant.
Antitumor activity of the combination of cDDP and metformin
Nude mice were transplanted subcutaneously with the different PDXs and were randomized when tumor weight reached ~150 mg. cDDP was given i.v. at the dose of 5 mg/kg q7 × 3, metformin was given orally (p.o.) at 400 mg/kg for 40 days (once daily for 40 days) in combination with cDDP (same schedule as single treatment). cDDP and metformin were dissolved respectively in Phosphate Buffer Saline (PBS) and in sterile water; control mice were treated with the same drug vehicles, following the same schedule.
Treatment evaluation
Mice were monitored twice a week; tumor growth was measured with a Vernier caliper, and tumor weight (mg = mm3) calculated as follows: [length (mm) × width 2 (mm2)]/2 and body weight was registered as indirect measure of drug toxicity. The efficacy of the treatment was expressed as best tumor growth inhibition [%T/C = (mean tumor weight of treated tumors/mean tumor weight of control tumors) × 100]. Statistical analysis of antitumor effect at the last day of observation was performed by one-way analysis of variance (ANOVA) test by GraphPad Prism v.6 software (GraphPad Software).
Metabolomic analysis
Metabolite extraction
For each xenograft, three different pieces (frozen tumor tissue samples, 20–50 mg) of the same tumor were taken from three different animals (n = 3) and homogenized using an Ultra Turrax (VWR, Pennsylvania, USA) with 10 µl/mg of extraction solvent (85:15 MeOH/H2O). The homogenized sample were stored at −80°C for 20 min and subsequently centrifuged for 15 min at 13,000g. Supernatants were collected and used for targeted and untargeted metabolomics analysis.
Untargeted metabolomics approach (FIA-QTOF-MS/MS)
Flow Injection Analysis/QTOF-Tandem Mass Spectrometry (FIA-QTOF-MS/MS) analysis was performed on an Agilent 1290 infinity Series coupled to an Agilent 6550 iFunnel Q-TOF mass spectrometer (Agilent, Santa Clara, CA, USA) equipped with an electrospray source operated in negative and positive mode. The flow rate was 150 μl/min of mobile phase consisting of isopropanol/water (60:40, v/v) buffered with 5 mmol/l ammonium at pH 9 for negative mode and methanol/water (60:40, v/v) with 0.1% formic acid at pH 3 for positive mode. Reference masses for internal calibration were used in continuous infusion during the analysis (m/z 121.050873, 922.009798 for positive and m/z 11.9856, 1033.9881 for negative ionization). Mass spectra were recorded from m/z 50 to 1100. Source temperature was set to 320°C with 15 l/min drying gas and a nebulizer pressure of 35 psig. Fragmentor, skimmer, and octopole voltages were set to 175, 65, and 750 V, respectively. Tandem mass spectrometry (MS/MS) fragmentation pattern of the significantly features were collected and used to confirm metabolite identity. Before each sample was run, a blank sample [isopropanol/water (60:40, v/v) negative, methanol/water (60:40, v/v) with 0.1% formic acid positive] to minimize the carry-over effect. This method allows a rapid metabolic profiling of polar and nonpolar compounds with the exclusion of lipid classes which were not considered in untargeted data elaboration due to the intrinsic method limitation in the discrimination of isobaric forms.
All steps of data processing and analysis were performed with MATLAB R2016a (MathWorks, Natick, MA, US) using in-house developed script following the workflow proposed by Fuhrer 18 . Centroid m/z lists were exported to .csv format. Briefly, in this procedure, we first subtract from each sample its relative blank sample to minimized the carry-over effect then, we applied a cutoff to filter peaks of less than 500 ion counts for negative and 1000 ion counts for positive ionization to avoid detection of features that are too low to be statistically significant. Centroid m/z lists from different samples were merged to a single matrix by binning the accurate centroid masses within the tolerance given by the instrument resolution (about 10 ppm). The output m × n matrix contains the m peak intensities of each mass for the n analyzed samples. Because mass axis calibration is applied online during acquisition, no m/z correction was applied during processing to correct for potential drifts. Output m/z list was submitted to statistical analysis (univariate pairwise comparison Mann–Whitney–Wilcoxon test, JMP pro12, SAS) in order to select features with a statistical significance between groups of comparisons. Significant altered features were identified by database searches (HMBD, http://www.hmdb.ca/; METLIN, http://metlin.scripps.edu) in positive and negative ionization, considering only protonate/deprotonate ion. Confirmed identifications were reported only for metabolites with accurate mass match <10 ppm and an MS/MS fragmentation patterns similarity > 99% relative to the reference compound present on the database.
Targeted metabolomics analysis
A targeted quantitative approach using a combined direct-flow injection and liquid chromatography tandem MS/MS assay (AbsoluteIDQ® p180 kit, Biocrates, Innsbruck, Austria) was applied as previously published published 19 . The method of AbsoluteIDQ® p180 kit conforms with the US Food and Drug Administration Guideline ‘Guidance for industry: bioanalytical method validation,’ which implies proof of reproducibility within a given error range. The method combines derivatization and extraction of analytes with the selective mass-spectrometric detection using multiple reaction-monitoring pairs. Isotope-labeled internal standards are integrated into the platform for metabolite absolute quantification. This strategy allows simultaneous quantification of 186 metabolites (40 amino acids and biogenic amines, 40 acylcarnitines, 90 glycerophospholipids, 15 sphingomyelins, 1 monosaccharide). The list of measurable metabolites using the Biocrates Absolute IDQ® p180 kit and their biological relevance is provided in Table S1. Significant metabolite changes were evaluated using univariate pairwise comparison Mann–Whitney–Wilcoxon test (JMP pro12, SAS).
Metabolic pathway analysis
For biological interpretation of the metabolite dataset, we mapped the significant metabolites derived from both untargeted and targeted approaches into the KEGG pathway database (Kyoto Encyclopedia of Genes and Genomes; www.genome.jp/kegg/), using MetaboAnalyst 3.0 (CA, USA), a comprehensive online tool suite for metabolomic data analysis and interpretation (www.metaboanalyst.ca). Enrichment analysis (EA) tools were used to identify metabolic pathways most likely to be associated with the cDDP acquired resistance. Differential abundance score was calculated for each significant enriched pathway as reported by Hakimi et al., 2016. 20
Gene expression analysis
Total messenger ribonucleic acid (mRNA) was extracted from cDDP-sensitive and cDDP-resistant xenograft snap-frozen samples for each xenograft by Maxwell technology (Promega, Madison, WI, USA). Tumor PDX samples were analyzed by real-time polymerase chain reaction (RT-PCR) to assess the percentage of murine deoxyribonucleic acid (DNA) contamination using primers specifically designed to distinguish human from murine actin. All the samples had a similar human actin content of more than 85%. The RT2 Profiler PCR Arrays (Qiagen, Hilden, Germany) are designed to analyze a panel of genes related to the glucose metabolism. For each plate, two DDP-S and two DDP-R samples of the same xenograft were included. We considered a fold of regulation of ⩾2 and ⩽−2 as significant up- and downregulation, respectively. 21 If a gene was found to be differentially regulated between sensitive and resistant samples only in one/two xenograft couple/s according to our parameters, we searched the value of fold regulation in the other couple/s. If the value of its fold regulation were be approximated to 2 or −2 values, we included that gene also in the analysis.
Real-time validation assays
For validation assays, total mRNA was retrotranscribed by RT2 First Strand kit (Qiagen). Next, gene expression was evaluated by RT-PCR with ad hoc-designed primers (Primer3, http://primer3.ut.ee/). Gene expression data were quantified through a calibration curve and were normalized by gene expression of a housekeeping gene (actin).
In vitro studies
Ovarian cancer PDXs were excised from mice at sacrifice. Tumors were mechanically disintegrated by scissors, and then enzymatically by collagenase (25,000 U/ml, Sigma-Aldrich, Saint Louis, MO, USA) at 37°C for 1 h. The cell suspension was filtered through a gauze and plated for 30 min in a Petri dish (Corning, Corning, NY, USA) in order to make fibroblast cells attach to the plastic surface.
Seahorse analysis
Seahorse provides accurate real-time measurements of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) at basal condition and after acute stress. The cell suspension (300,000 cell/ml) was plated for 30 min in tissue-treated Petri for 30 min as a cleaning passage, and the surnatant was counted with a Burker camera (Prodotti Gianni srl. MI, Italy). Cells were then seeded at a concentration of 40,000 cells/well in a Seahorse cartridge (Agilent Technologies), then the Mito Stress and the glycolysis stress tests were performed as specified in the manufacturer’s protocols (Agilent technologies). Three replicates were performed for each group. Statistical analysis was performed by Bonferroni multiple comparison test (GraphPad Prism v6).
Results
Isolation of ovarian cancer PDXs with acquired resistance to cisplatin
We recently obtained a panel of PDXs from ovarian human samples that well represent the heterogeneity of human tumor in their morphology, molecular profile and pharmacological response. 22 Among them, we selected three high-grade models (MNHOC124, MNHOC124LP, and MNHOC239, from now referred to as #124, #124LP, and #239) responsive to cisplatin (cDDP) treatment (sensitive, S) in order to obtain cDDP-resistant (R) models [Figure 1(a)]. #124 is a mixed serous/endometrioid histotype carcinoma; #124LP is a subline obtained from #124 that has been passaged for nine passages in vivo; and #239 is a serous carcinoma. All the xenografts are high-grade tumors and TP53 mutated (Table S2). As already reported for #124 and #239,22,23 cDDP treatment was able to induce tumor regressions and was associated with a striking antitumor activity as indicated by the T/C% values. We obtained sublines resistant to cDDP after in vivo drug treatment of mice bearing sensitive tumors, as specified in Materials and Methods. After a total of five to seven in vivo cDDP treatment cycles, we obtained three PDX models resistant to the drug (R). Indeed, no tumor regressions or stabilizations were observed in resistant xenografts after cDDP treatment, and an increase in the T/C% values was observed [Figure 1], indicative of loss of cDDP activity [Figure 1(b–d)]. Histological analysis indicates no change in histotype after treatment with cDDP (data not shown). #124-R and #124LP-R, but not #239-R sublines, displayed a statistically significant increase in tumor growth as suggested by a decreased median time to reach 1gr (median of 32.8 and 30.9 days to reach 1 gr, respectively) compared with the corresponding S xenografts (median of 47.8 and 42.2 days to reach 1 gr, respectively; Table S2).

cDDP antitumor activity in the different PDXs.
Expression of genes belonging to the glycogen, glycolysis and TCA cycle pathways in cDDP-sensitive and -resistant PDXs
We use these experimental settings to investigate whether tumors from S- and R-PDXs exhibited a different metabolic asset. We first investigated the expression of genes coding for key metabolic enzymes or regulators of glucose and glycogen metabolism, pentose phosphate pathway and tricarboxylic acid (TCA) cycle.
A similar number of deregulated genes in terms of fold regulation between resistant and sensitive xenografts (#124 n = 14; #124LP n = 12; #239 n = 13; see Material and Method section) was observed [Figure S1(a)] in the expression of 85 genes among sensitive and resistant xenografts in the three different couples. The genes differentially expressed in the three xenografts are listed in Figure 2(a) and are genes coding enzymes of the glycolytic pathway, followed by TCA and glycogen pathways. Of note, the pentose phosphate pathway was only marginally affected. We validated, by RT-PCRs with ad hoc-designed primers, the genes altered in two out of three PDX couples, and for most of the genes, the expression trend identified in the PCR profiler assay was confirmed [Figure 2(b)]. Indeed, we corroborated the statistically significant upregulation of IDH2 (isocitrate dehydrogenase 2) and PYGL (glycogen phosphorylase), and the downregulation of PDK3 (pyruvate dehydrogenase 3) in the corresponding resistant xenografts. ALDOC (aldolase fructose-bisphosphate C) expression was found to be statistically upregulated only in #124LP-R and #239-R, while only a trend was observed in the #124 couple. RBKS and MDH1B were validated in downregulation of #124-R and #239-R versus -S xenografts. On the contrary, the downregulation of PGK2 was not validated [Figure S2(a)]. The RT2 Profiler PCR array did not include the monocarboxylate and glutamine transporters such as lactate transporters MCT1 and MCT4 (SLC16A1, and SLC16A4), the glucose transporter GLUT1 (SLC2A1), and the glutamine transporter ASCT2 (SLC1A5), all involved in the regulation of glycolysis. We studied their gene expression levels and found that SLC16A4 and SLC1A5 were upregulated in two out of the three DDP-R xenografts [Figure S2(b)]. SLC2A1 and SLC16A1 were marginally altered [Figure S2(b)]. No modification of MCT4 expression between resistant and sensitive tumor samples was found by immunohistochemistry (data not shown). These findings indicate a lack of change in the expression of genes regulating glycolysis in resistant tumor samples.

Genes differentially expressed in sensitive and resistant ovarian cancer PDXs.
cDDP-resistant PDXs display a different metabolic layout
We used an integrative mass spectrometry-based metabolomic approach, combining targeted (T) and untargeted (UT) strategies, to increase the metabolome coverage, thus providing a wider perspective of the tumor metabolic pathways changes occurring in each sensitive and the corresponding resistant xenografts (#124, #124LP, #239, three biological replicates for each PDX,n = 3). OPLS-DA (orthogonal projections to latent structures discriminant analysis) reveals the presence of metabolic features able to segregate the three sensitive from the relative cDDP-resistant PDXs [Figure 3(a)]. A closer segregation could be observed among #124- and #124LP-sensitive samples suggesting that both the in vivo passages do not greatly alter tumor metabolic layout and the in vivo cDDP treatment induces comparable metabolic changes.

Metabolic changes in the R-PDXs.
To identify metabolites associated with cDDP resistance, we used pairwise comparison (Mann–Whitney p < 0.05) in resistance relative to their sensitive counterparts. We found 72 (48 T, 24 UT), 46 (16T, 27 UT), 32 (10T, 22UT) significantly deregulated metabolites respectively in #124, #124LP, #239-resistant relative to their sensitive counterparts (Tables S3, S4 and S5). #124-R PDX showed the highest number of deregulated metabolites, followed by #124LP-R and #239-R PDXs [40 versus 10 versus 9, Figure S1(b)]. Targeted metabolomics reveal a significant reduction in the levels of glycerophospholipids and sphingomyelins in the #124 DDP-R xenograft only, while #124LP-R and #239-R showed only a marginal alteration of lipid profile, mainly related to lysophosphatidylcholine and sphingolipid species (Tables S2, S3, and S4). Eleven metabolites were found commonly altered in all the cDDP-resistant xenografts [Figure S1(b)]. Metabolic EA (MetaboAnalyst), using all (from T and UT metabolomics) the significant deregulated metabolites in each xenograft pair (Tables S3, S4, S5), highlighted the TCA and urea cycle as the most enriched pathways (p < 0.05, FDR < 0.05) [Figure 3b–d)]. Among the metabolites belonging to the TCA pathway, only pyruvic acid and fumarate showed a comparable and consistent downregulation in all the R-PDXs as compared with the corresponding S-PDXs (Figure 4 and Tables S3, S4, S5). We observed a consistent, even if different, deregulation of urea cycle metabolites in the three R- and S-PDX couples (Figure 4 and Table S6). When we investigated the expression of genes coding the key enzymes of the urea cycle, xenograft #124 showed a significant alteration in gene coding for ornithine transcarbamylase (OTC) and arginosuccinate synthase (ASS1) associated with a general deregulation of all metabolites of the cycle (Figure S3). #124LP-R showed the significant upregulation of ARG1 gene and of ASS1 associated with a significant lower level of its metabolic substrate citrulline (Figure S3). Interestingly, #239-R was the most deregulated xenograft with significant alteration of all genes belonging to the urea cycle (OTC, ASS1, ASL and ARG1), although no relevant alterations were found with associated metabolites (Table S6). The urea cycle, important for the synthesis of nitrogen-containing compounds, also fuels the polyamine metabolism through the generation of ornithine. Significant decreased levels of all polyamines (putrescine, spermidine, spermine) were found in #239-R xenograft compared with its sensitive counterpart, whereas they increased (although not always significantly) in both #124-R and #124LP-R (Figure 4).

Specific metabolic alterations between resistance and sensitivity in each PDX couple (#124, #124LP, #239).
Within each pair of xenografts, we observed alterations in specific biochemical pathways interconnected with the above over-represented pathways (i.e. the TCA and urea cycle) in resistant xenografts. In particular, we observed enriched arginine and proline metabolism in #124-R, alanine metabolism in #124LP-R, alanine metabolism, malate–aspartate shuttle, ammonia recycling and gluconeogenesis in #239-R. Interestingly, alanine metabolism was significantly over-represented in both #124LP-R and #239-R [Figure 3(b–d)].
Different ability to respond to energy demand between resistant (R) and sensitive (S) xenografts
As a whole, the expression of metabolic genes and metabolic profile supports a perturbation of the glycolytic axis, that is, increased PYGL and ALDOC mRNA expression, to fuel the TCA cycle and a sustained mitochondrial respiration in R-PDXs. This prompted us to perform functional experiments with Seahorse technology to measure in RT the OCR and the extracellular acidification rate (ECAR) to indirectly explore the mitochondrial and glycolytic functions; in fact, OCR is an indicator of mitochondrial respiration, and ECAR is largely the result of glycolysis. Cell suspensions obtained from the digestion of fresh #124LP tumors (S and R) were processed, as detailed in Materials and Methods, and after 48 h, cells underwent acute stress stimuli. The #124LP-R cells showed higher adenosine triphosphate (ATP) production in basal conditions [Figure 5(a), right] as compared with cells derived from #124LP-S xenografts. When cells underwent acute stress (treatment with oligomycin, a complex V inhibitor) with p-trifluoromethoxyphenylhydrazone, a protonophore, and lastly with antimycin A and rotenone, (inhibitors of complex III and I) #124LP-R cells showed stronger ability to respond to an energetic demand, and a higher rate of respiration than S cells [Figure 5(a), right]. Similar results were obtained with #124 xenograft pair [Figure 5(c)]. We then measured glycolysis and glycolytic capacity to calculate the glycolytic reserve and nonglycolytic acidification in both #124LP-S and -R xenografts by evaluating the ECAR in response to a glycolytic stress. We did not find any differences between “Raud S PDXs” [Figure 5(b)] in line with results of RT-PCR and immunohistochemistry (IHC) analysis.

Metabolic measurements in the #124LP and #124 PDX pair.
Metformin treatment reverses cDDP resistance in vivo
Based on the data obtained from metabolomic analysis, gene expression, and functional assays, we hypothesized that resistant xenografts have an increased mitochondrial activity compared with S xenografts. To test if cDDP resistance could be reversed by interfering with the increased mitochondrial activity, we tested the combination of cDDP and metformin (a drug able to interfere with mitochondrial function) in vivo. We transplanted #239-R and when tumor masses reached about 150 mg, mice were randomized to receive vehicle, cDDP or a combination with cDDP and metformin. As shown in Figure 6(a), the addition of metformin increased cDDP antitumor activity. At this dose (400 mg/kg daily for 40 days) metformin does not exert any antitumor activity (data not shown) and it was able to activate adenosine monophosphate kinase (AMPK; Figure S4). Indeed, even if two out of eight mice in the group treated with the combo were sacrificed for tumor burden at day 50, at the last day of observation (day 62), mean values of tumor weight of the combination and of the single cDDP-treated groups were statistically different [Figure 6(b)]. Similar results were obtained when treating mice bearing #124-R tumors, even if in this case no statistical significance was reached, likely due to the lower number of mice used [Figure 6(c, d)].
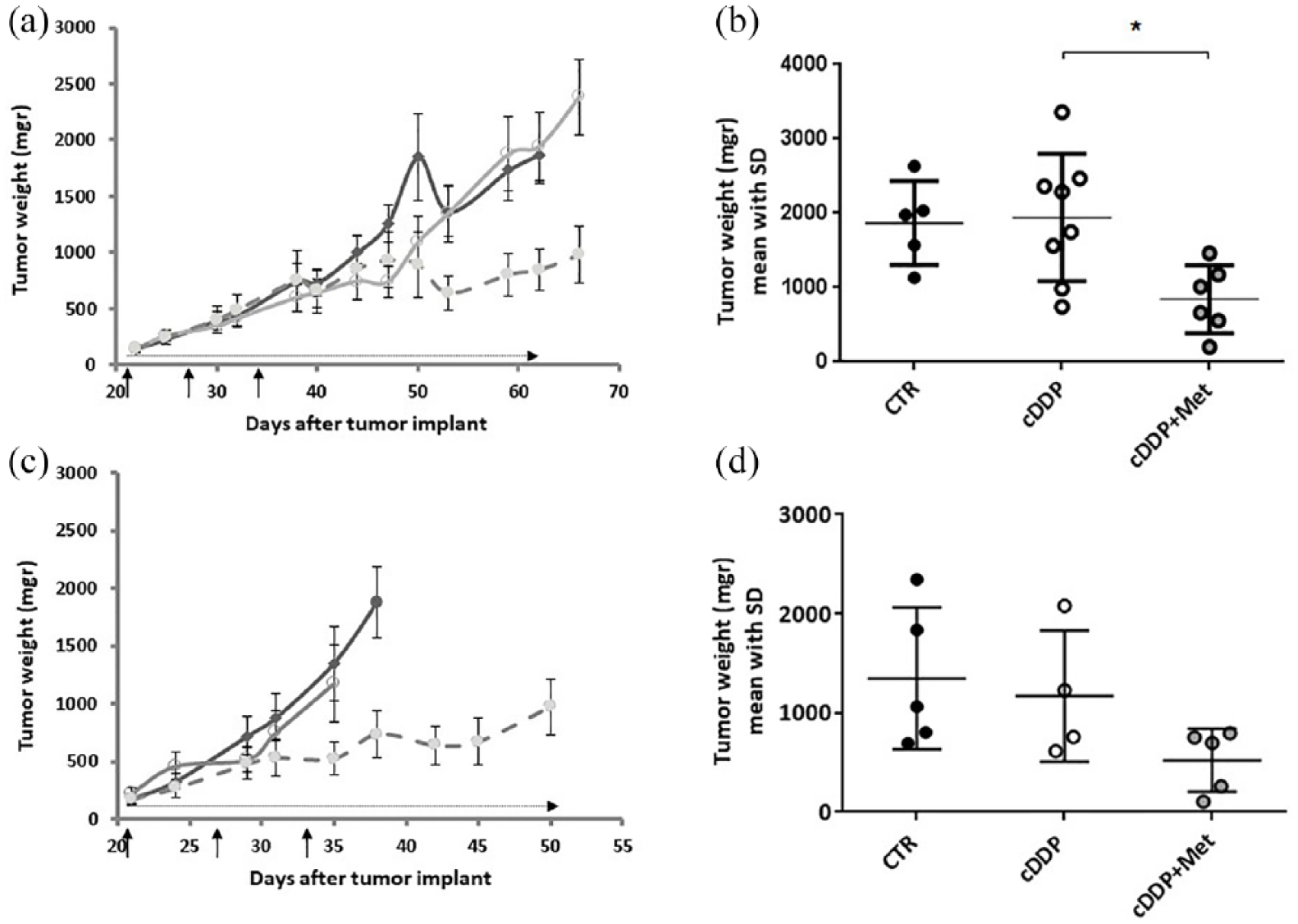
cDDP and metformin treatment in ovarian cancer PDXs.
Discussion
EOC is often initially responsive to a first-line platinum-based treatment (~70% of patients respond to therapy), but unfortunately, most of the patients will relapse with platinum-resistant disease. The development of resistance to a platinum therapy is an important issue, as it represents one of the causes of poor prognosis of these patients (5-year survival of less than 30% in most cases). With this work we have raised awareness of the possible mechanisms for acquired resistance to cDDP and suggested new therapeutic interventions.
In particular: (a) we obtained three new PDX models of acquired cDDP resistance after in vivo treatment; (b) we found, through metabolomic and gene expression approaches, that glycolysis, TCA and urea cycle pathways were deregulated in R- versus S-PDXs; (c) we observed that OCR and mitochondrial respiration were higher in R-PDXs than in S-PDXs under acute stress conditions; and (d) we proved that metformin, a drug able to inhibit the mitochondrial activity, was able to partially reverse cDDP resistance in vivo.
The acquisition of therapy resistance has been recently associated with metabolic switching in different tumors, including ovarian carcinomas.13–15,22–28 However, the majority of these results have been obtained using cancer cell lines or cell-line-derived xenografts, 12 and very few studies have been carried out using in vivo models that represent the clinical setting. 29
We have recently established an ovarian PDX xenobank that reproduces the complexity and heterogeneity of human ovarian carcinoma. 22 Starting from three high-grade ovarian carcinomas, we obtained, by in vivo cDDP treatment, three sublines with acquired resistance to cDDP. In this study, they have been used to investigate the possible role of metabolic rewiring in the resistance to cDDP. Even though a different metabolic profile has been reported between sensitive and resistant ovarian cell lines in vitro, 12 this is the first report that applies a multilevel pipeline (MS-based metabolomics and metabolic gene expression profiling) on ovarian PDXs made resistant in vivo to cDDP.
Despite the heterogeneity of metabolic responses after acquisition of platinum resistance among the PDXs, our multilayer strategy pointed toward major metabolic alterations in glycolysis, TCA and urea cycle biochemical routes in all the resistant PDXs relative to sensitive counterparts. We found an induction of glycolytic genes and a concomitant downregulation of the gluconeogenic axis counterpart, associated with decreased pyruvate level and unchanged lactate production. The increase in the glycolytic genes was combined with the induction of the PYGL. The glycogen degradation by PYGL is a source of glucose-6 phosphate, which can be used not only to sustain glycolytic reactions, but also to fuel the pentose phosphate pathway, providing the synthesis of nucleotides and reduced Nicotinamide Adenine Dinucleotide Phosphate (NADPH). 30
Together, these findings suggest the presence of an enhanced glycolytic pathway in the cDDP-resistant sublines that seems to fuel oxidative phosphorylation, rather than aerobic glycolysis, to support the bioenergetic function. In concordance with this hypothesis, we observed a reduction of pyruvate dehydrogenase kinase 3 (PDK3), which plays a critical role in the control of the glycolytic–mitochondrial axis. It has been reported that PDK3 knockdown indeed promotes the oxidative decarboxylation of pyruvate to produce acetyl coenzyme A and Nicotinamide Adenine Dinucleotide (NADH) to fuel the TCA pathway and mitochondrial respiration. 31 Although the TCA deregulation among the different resistant xenografts was heterogeneous, the alterations of both TCA genes and related metabolites in cDDP-resistant xenografts were coherent with the presence of perturbed mitochondrial functions. We observed that all resistant xenografts displayed a common decrease in levels of fumarate and increased isocitrate dehydrogenase 2 (IDH2), both supporting a more processive TCA and enhanced mitochondrial respiration. IDH2, along with IDH1, is a key metabolic enzyme that converts isocitrate to α-ketoglutarate and is frequently mutated in different cancers, 32 and the mutation is associated with an enzymatic gain of function. 33 While no mutations have been reported in ovarian carcinoma, 1.9% of the The Cancer Genome Atlas (TCGA) cases displayed IDH2 amplification. However, when we looked for correlation, no association between IDH2 expression levels and patient overall survival in the TGCA data set could be found (data not shown).
Resistant PDXs showed a perturbation of genes and metabolites belonging to the urea cycle, even though a common trend of deregulation could not be found. Acute cDDP treatment in pluripotent stem cells has been shown to induce modification in metabolites and enzymes related to the urea cycle; 34 however, no data are available on multiple cDDP treatments leading to drug resistance. We found a common lower level of fumarate in all the R-PDXs. At the biochemical level, fumarate is the metabolic link between the TCA and urea cycle where, during the generation of arginine, fumarate is generated as a byproduct; these data would again suggest a more processive TCA and urea cycle pathway. The urea cycle is not only important for the synthesis of nitrogen-containing compounds, but also for providing polyamines, which are small aliphatic polycations ubiquitously present in cells. It is known that polyamines bind to DNA, and modify its secondary structure, including chromatin condensation and DNA-matrix association.35,36 However, even if we found modulation of polyamine levels (putrescine, spermidine, and spermine) in our resistant models, the deregulation was different (upregulation and downregulation) in the three resistant PDXs, both suggesting an unrelated effect or specific underlying cDDP-resistant associated mechanisms.
Overall, our metabolic profiling suggests an adaptive oxidative phosphorylation in cDDP-resistant ovarian PDXs to sustain growth. Such adaptation partially corroborates our recent findings,24,37,38 in which ovarian cancer cells derived from ascites of nonresponding patients are much less sensitive to glucose deprivation in vitro than cells derived from platinum-responding patients. This metabolic snapshot was also functionally confirmed by the increased OCR and ATP and lower ECARs in primary resistant cultures as compared with sensitive culture derived from the corresponding PDXs. These findings agree and support other work that suggested the association of the resistance to cDDP with an increased oxidative metabolism,24,38 and lead us to hypothesize that its interference could be of therapeutic value. As our data support support this hypothesis, we tested whether metformin, the most commonly prescribed drug for type II diabetes, able to affect both complex I and ATP synthase in mammalian mitochondria, could resensitize resistant tumors to cDDP. Cotreatment of metformin and cDDP could partially reverse cDDP resistance in vivo, as suggested by the lower mean resistant tumor weights of mice treated with the combination than single cDDP-treated mice. It can be hypothesized that chronic treatment with metformin inhibits mitochondrial metabolism reversing the tumor metabolic properties to that of cDDP-sensitive PDX with a regain of drug sensitivity. Metformin has been shown to have pleotropic effects in normal and cancer cells, including interference with the mitochondrial complex I, with derangement of the AMP/ATP balance and activation of AMPK, described as one of the central regulators of cell growth and metabolism. 39 As for any drug, metformin in vivo effects largely depend on the levels reached in plasma and tumor tissue. Dowling and colleagues 40 have shown that a dose of 5 mg/ml in drinking water for 16 days or intraperitoneal dose of 125 mg/kg achieved, respectively, an average tumor concentration of 32µmol/l (range 9.1–55.7 µmol/l) and 77 µmol/l (range 41.6–99.0 µmol/l), doses able to activate AMPK. The dose of 400 mg/kg given per os for 40 days should enable achieving active drug tumor concentration in tumors of mice treated with this dose, as demonstrated by the activation of AMPK (Figure S4). However, considering the pleiotropic effect of metformin on cells, as recently reviewed,41,42 we cannot rule that other nonmetabolic mechanisms are occurring. It has been reported that cancer stem cells are enriched in resistant tumors and that metformin preferentially kills cancer stem cells. 43 This hypothesis is, however, challenging to test in our experimental setting, as the phenotypical and functional traits of stem cells in ovarian cancer are still elusive. 44 To add to the variety and complexity of in vivo metformin effects, very recently, metformin has been found to repress the cDDP-stimulated interleukin 6 expression in ovarian cancer tumor stroma, reported to be associated with cDDP resistance 45 ; in addition, Li and colleagues showed metformin treatment’s ability to block the suppressive function of myeloid-derived suppressor cells in ovarian cancer patients. 46
Conclusion
The data herein presented strongly reinforce the idea that the development of acquired cDDP resistance in ovarian cancer can be associated with a rewiring of tumor metabolism and this can be exploited therapeutically.
Supplemental Material
Suppl_Fig_1 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Fig_1 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Fig_2 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Fig_2 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Fig_3 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Fig_3 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Fig_4 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Fig_4 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Methods – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Methods for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Table_1 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Table_1 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Table_2 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Table_2 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Table_3 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Table_3 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Table_4 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Table_4 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Table_5 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Table_5 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Supplemental Material
Suppl_Table_6 – Supplemental material for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts
Supplemental material, Suppl_Table_6 for Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts by Francesca Ricci, Laura Brunelli, Roberta Affatato, Rosaria Chilà, Martina Verza, Stefano Indraccolo, Francesca Falcetta, Maddalena Fratelli, Robert Fruscio, Roberta Pastorelli and Giovanna Damia in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We are gratefully acknowledged to Judith Bagott for editing the manuscript and to Associazione Italiana per la Ricerca sul Cancro (AIRC) for financial support.
Francesca Ricci and Laura Brunelli contributed equally to this work. Roberta Pastorelli and Giovanna Damia contributed equally to this work
FR, LB, RP and GD conceived the study and participated in the study design, performance, coordination and manuscript writing. RF, LB, RA, RC, MV, SI, FF, MF and FR performed the research. All authors have read and approved the final manuscript
Funding
The generous contribution of the Italian Association for Cancer Research (AIRC) is gratefully acknowledged (IG19797 to GD and IG18803 to SI).
Conflict of interest statement
The authors declare that there is no conflict of interest.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.