
Abstract
Objective:
To determine the efficacy and safety of intermittent docetaxel chemotherapy guided by circulating methylated glutathione S-transferase Pi-1 (mGSTP1) in men with metastatic castration-resistant prostate cancer (CRPC).
Patients and Methods:
GUIDE (NCT04918810) is a randomised, two-arm, non-comparative phase-2 trial recruiting 120 patients at six Australian centres. Patients with Prostate Cancer Working Group-3 defined metastatic CRPC who are commencing docetaxel 75 mg/m2 q3w will be pre-screened for detectable mGSTP1 at baseline ± following two cycles of treatment. Those with detectable plasma mGSTP1 at baseline that becomes undetectable after two cycles of chemotherapy will be eligible for GUIDE. Prior to Cycle 4 of docetaxel, these patients are randomised 2:1 to one of two treatment arms: Arm A (cease docetaxel and reinstitute if mGSTP1 becomes detectable) or Arm B (continue docetaxel 75 mg/m2 q3w in accordance with clinician’s usual practice). The primary endpoint is radiographic progression-free survival. Secondary endpoints include time on treatment holidays, safety, patient-reported outcomes, overall survival, health resource use, and cost associated with treatment. Enrolment commenced November 2021.
Results and Conclusion:
The results of this trial will generate data on the clinical utility of mGSTP1 as a novel biomarker to guide treatment de-escalation in metastatic CRPC.
Keywords
Background
Docetaxel represents a key treatment option for metastatic prostate cancer, offering improvements in median overall survival (OS), cancer-related symptoms, and quality of life when administered in metastatic hormone-sensitive prostate cancer (mHSPC)1,2 or castrate-resistant prostate cancer (mCRPC) 3 settings. Despite optimisation, docetaxel is associated with significant fatigue, peripheral neuropathy, and neutropaenia, which impact tolerability particularly for patients who may be older and often with multiple co-morbidities1,3,4 Biomarker-driven de-escalation of treatment may allow improved balance of cancer control and quality of life against toxicity for people with incurable prostate cancer.
Conventional biomarkers in this space are flawed, leaving space for novel approaches including methylated glutathione S-transferase Pi-1 (mGSTP1) to guide treatment de-escalation.
mGSTP1 as a biomarker
The GSTP1 gene encodes for the GSTP1 enzyme that has an important role in normal metabolism, detoxification, and elimination of potentially genotoxic stressors, which helps to mitigate deoxyribonucleic acid (DNA) damage alongside other glutathione S-transferases. Notably, GSTP1 also aids in the metabolism of some cytotoxic agents, including platinum-based chemotherapy such as cisplatin and carboplatin, and differences in expression of GSTP1 when evaluated in a subset of ovarian cancers appeared to impact response to treatment. 5
GSTP1 expression is epigenetically reduced in >90% of all prostate cancers.6,7 This occurs secondary to aberrant DNA methylation of its CpG island and first exon. Owing to its prevalence, serum-free methylated GSTP1 (mGSTP1) is more specific for prostate cancer than prostate-specific antigen (PSA) 8 and importantly, is not observed in normal prostate 9 or non-prostate tissue, 10 making it an attractive biomarker for use in de-escalation studies.
In early clinical studies, mGSTP1 was reliably detected in serum and urine of individuals with prostate cancer 11 and associated with biochemical relapse if present in pre-operative serum samples of individuals with localised prostate cancer. 12 In larger data sets of men with mCRPC, levels of expression of circulating mGSTP1 were prognostic in discovery and internal validation cohorts of men with mCRPC. Detectable baseline plasma mGSTP1 circulating tumour DNA (ctDNA) was associated with inferior OS in both the discovery and validation cohorts, even in the absence of treatment. In men receiving chemotherapy, a decline in mGSTP1 ctDNA after one cycle of chemotherapy corresponded with PSA response and improved OS. 13
A post hoc analysis of a large phase-3 trial of men with mCRPC (SYNERGY) subsequently showed that circulating mGSTP1 is detectable in most men at baseline (81%) and becomes undetectable in around half of those men during treatment (53% after two cycles of docetaxel). 14 Undetectable mGSTP1 after two cycles of treatment was associated with longer OS (hazard ratio (HR) 0.36, 95% confidence interval (CI) 0.29–0.46; p < 0.00001) and longer time to PSA progression (HR 0.44, 95% CI 0.35–0.56; p < 0.00001), than persisting detectable levels. These observations are key to the design of the GUIDE clinical trial.
Historically, treatment de-escalation in mCRPC guided by PSA was evaluated.15 –18 Most focused on ‘treatment holidays’ where chemotherapy was temporarily withheld following a ‘significant’ reduction in PSA and re-commenced following a ‘significant’ rise. However, due to the inherent inaccuracies of PSA, the definition of a significant PSA change varied between studies and was necessarily stringent, allowing only a minority of patients to participate. Reassuringly, this approach did allow prolonged treatment holidays of a median 18–38 weeks in these studies and offered an acceptable median radiographic progression-free survival (rPFS) of 12–17.5 months.15 –18 Intermittent dosing in this way may also delay the emergence of chemotherapy-resistant cell populations; however, clinical studies have been hampered by inadequate diagnostic tools to measure treatment response. 19 Improved and more accurate biomarkers such as mGSTP1 have the potential to better guide optimal treatment decisions.
Patient-related outcomes
Patient-related outcomes (PROs) have been integrated into multiple trials evaluating the benefit of treatment in metastatic prostate cancer. As docetaxel was established as a new standard of care in mCRPC, improvements in cancer-related symptoms and quality of life were observed 3 using the Functional Assessment of Cancer Therapy – Prostate (FACT-P) questionnaire. Modern studies have used additional instruments to evaluate specific toxicities and symptoms, such as pain and fatigue, that men undergoing treatment for prostate cancer may experience. 20 Increasingly, PROs are submitted alongside efficacy data to support regulatory approval of new therapeutic agents or regimens. 21
GUIDE: mGSTP1 utility for intermittent docetaxel therapy for metastatic prostate cancer clinical trial overview
GUIDE is a randomised, two-arm, non-comparative phase-2 trial, designed to determine the efficacy and safety of intermittent docetaxel chemotherapy guided by plasma mGSTP1 in men with metastatic CRPC. Eligible participants will be randomised in a 2:1 ratio to intermittent docetaxel therapy or continuous docetaxel (standard of care), provided they had detectable mGSTP1 at baseline that has converted to an undetectable mGSTP1 after two cycles of docetaxel chemotherapy (see Figure 1 for study schema).
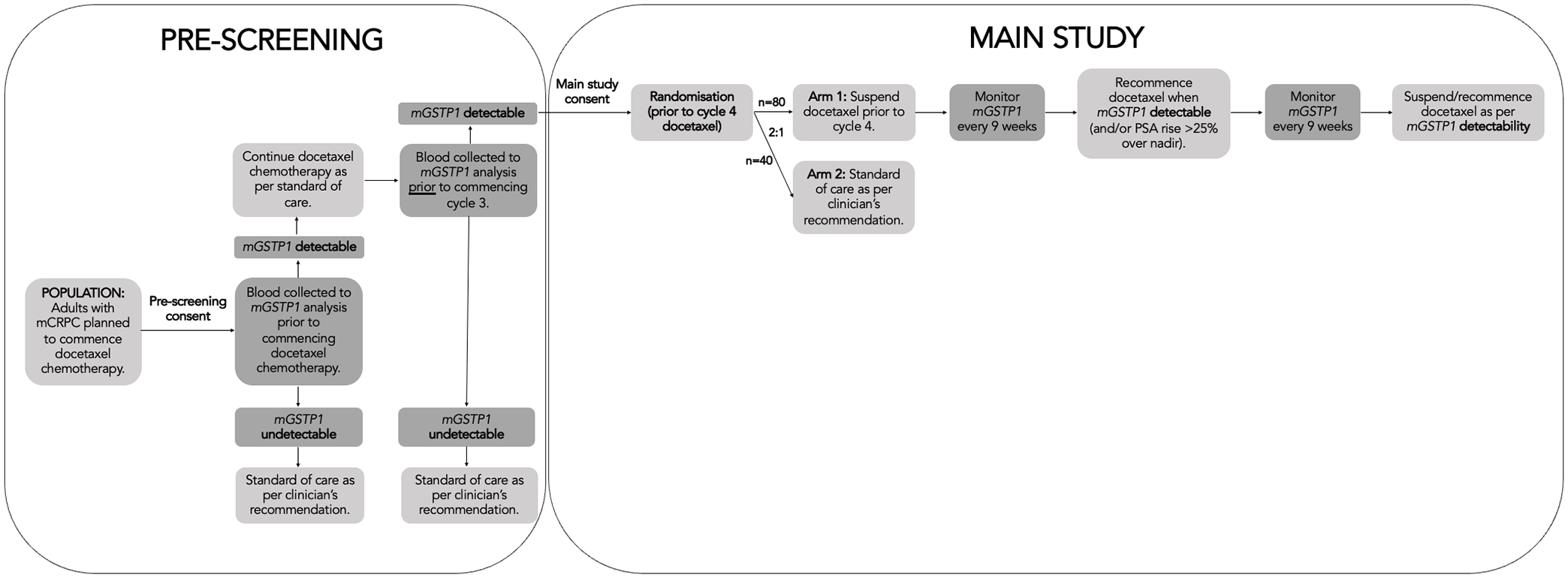
Study schema.
This trial is sponsored by the Australian and New Zealand Urogenital and Prostate (ANZUP) Cancer Trials Group, with funding from ANZUP Discretionary Funding Initiative, ANZUP Below the Belt Research Fund, Chris O’Brien Lifehouse Philanthropic Fund, the Twin Towns Foundation, the Cancer Institute of NSW, and the National Health and Medical Research Council. The trial was first registered at clinicaltrials.gov (NCT04918810) on 9 June 2021. Central ethical approval has been obtained from St Vincent’s Hospital Human Research Ethics Committee (HREC 2021/ETH00523) in 2021. Local ethical and governance approval has been or will be obtained for six participating Australian sites. The study is being conducted in accordance with the Declaration of Helsinki, 22 Note for Guidance on Good Clinical Practice, 23 and in compliance with the applicable laws and regulations including the NHMRC National Statement on Ethical Conduct in Human Research, 24 and the NHMRC Australian Code for Responsible Conduct of Research. 25 All participants will provide written informed consent.
The primary objective of this study is to evaluate the treatment outcomes of intermittent docetaxel in men with undetectable plasma mGSTP1 after two cycles of therapy for metastatic CRPC. The primary endpoint is rPFS, defined as investigator-assessed progression by Prostate Cancer Working Group 3 (PCWG3; for bone lesions) and Response Evaluation Criteria In Solid Tumours (RECIST) version 1.1 (for soft tissue lesions), or death due to any cause. The secondary objectives and endpoints are to describe/explore:
Duration and number of treatment holidays prior to permanent treatment discontinuation.
Safety: frequency and severity of adverse events (AEs) using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 5.
PRO.
OS: death from any cause.
Proportion of patients who could have had a treatment holiday based on changes in PSA level alone.
Health resource use and cost.
Methods
GUIDE’s target population is men with metastatic CRPC, defined by PCWG3, who are planned to commence docetaxel chemotherapy. Prior docetaxel is allowed if administered in the mHSPC setting and at least 2 years prior to study enrolment. The inclusion and exclusion criteria for pre-screening and the main study are outlined in Table 1.
Inclusion and exclusion criteria.

mGSTP1, methylated glutathione S-transferase Pi 1; RECIST, Response Evaluation Criteria in Solid Tumours
After providing consent for pre-screening, all patients will undergo plasma mGSTP1 analysis to assess preliminary eligibility for the main study. Those with baseline undetectable mGSTP1 will not be eligible and will return to usual care. Patients with baseline detectable mGSTP1 will commence docetaxel 75 mg/m2 intravenously every 21 days, with prednisolone orally 5–10 mg daily. After two cycles, repeat mGSTP1 analysis will be performed, and a third cycle of docetaxel will be administered, while repeat results are awaited. If patients have persistent detectable mGSTP1 at this time point, they will not be eligible for the main study and will return to usual care. Alternatively, if mGSTP1 becomes undetectable after two cycles of docetaxel, the patient will be invited to consent for the main study. Participants eligible for the main study are randomised in a 2:1 ratio to Arm A and Arm B, respectively.
The experimental group (arm A) will pause docetaxel after three cycles and undergo serial mGSTP1 analysis every 9 weeks and PSA analysis every 3 weeks, until mGSTP1 becomes detectable, at which time docetaxel and prednisolone will be re-instituted. At their discretion, clinicians may recommence docetaxel and prednisolone if serum PSA rises >25% above nadir before mGSTP1 becomes detectable. Subsequent treatment holidays are permitted in arm A if mGSTP1 becomes undetectable again. Prednisolone can be continued or ceased during the treatment holiday at the discretion of the treating clinician. Treatment will continue until disease progression, defined by RECIST v1.1 or PCWG3, or unacceptable toxicity. While measured serially during the study, PSA decline will not be used to initiate a treatment holiday; however, a PSA fall of >50% from pre-treatment levels (and absolute PSA level of <4 ng/mL) will be used to address this research question and secondary endpoint.
The standard of care group (arm B) will receive docetaxel in accordance with their treating clinician’s usual practice and/or local guidelines, until disease progression, defined by RECIST v1.1 or PCWG3, or unacceptable toxicity.
Dose delays and modifications
Upfront dose reduction of docetaxel is permitted at the discretion of the treating clinician. Dose delays and reductions are allowed for toxicity according to clinician’s usual practice. In general, docetaxel should be delayed during clinically relevant AEs of grade 3–4 severity and not restarted until the AE has resolved to grade 0–1. Docetaxel can be delayed a maximum of 14 days, after which, treatment should be discontinued permanently if grade >1 AEs persist. If multiple AEs occur and mandate dose reduction, the modification that results in the longest delay and lowest dose should be used.
Treatment discontinuation
Reasons for treatment discontinuation include disease progression, unacceptable toxicity, requirement for palliative radiotherapy required use of a prohibited therapy, withdrawal of consent, non-compliance, or other reasons in the opinion of the investigator that continuing the study is either no longer safe, or no longer in the best interest of the patient.
Assessments
Clinical assessment, including serum PSA, will be conducted at baseline and every 3 weeks until permanent cessation of docetaxel. Patients in arm A and on a treatment holiday, must continue the same schedule, until disease progression defined by RECIST v1.1 or PCWG3, or unacceptable toxicity. After permanent treatment cessation (end-of-treatment visit), patients will undergo PSA evaluation every 3 months for 12 months, and then every 6 months until disease progression. Survival follow-up will continue thereafter every 3 months after disease progression until the last patient completes 2 years of follow-up. Computerised tomography (CT) or magnetic resonance imaging (MRI) of chest, abdomen, and pelvis, and whole-body bone scan (WBBS) will be performed during pre-screening, and then every 12 weeks during treatment until disease progression by RECIST v1.1 or PCWG3. PROs will be collected at screening for the main study and then every 12 weeks until disease progression defined. Patients in arm A and on a treatment holiday must continue the same schedule. The annotated schedule of assessments is provided in Table 2.
Schedule of assessments.
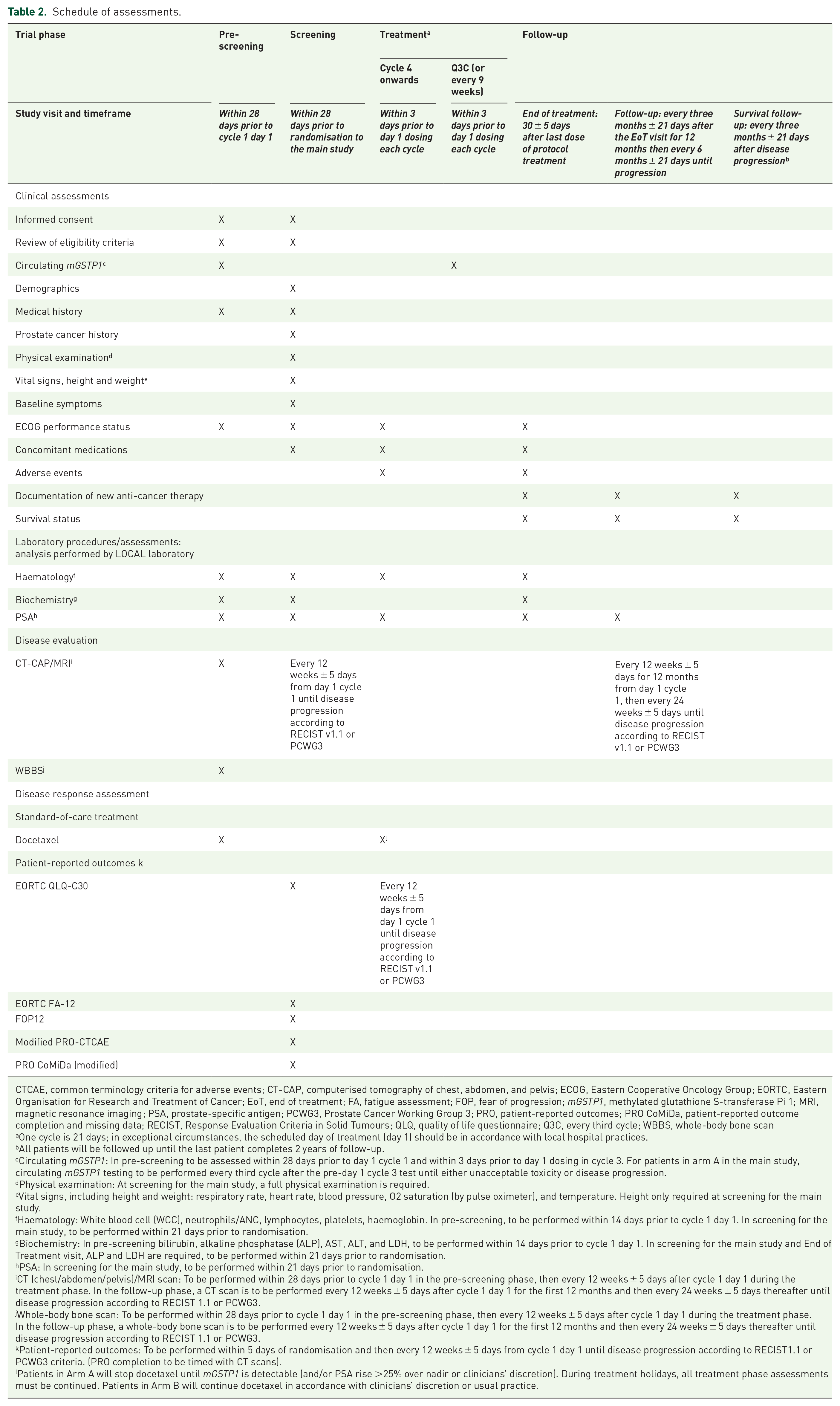
CTCAE, common terminology criteria for adverse events; CT-CAP, computerised tomography of chest, abdomen, and pelvis; ECOG, Eastern Cooperative Oncology Group; EORTC, Eastern Organisation for Research and Treatment of Cancer; EoT, end of treatment; FA, fatigue assessment; FOP, fear of progression; mGSTP1, methylated glutathione S-transferase Pi 1; MRI, magnetic resonance imaging; PSA, prostate-specific antigen; PCWG3, Prostate Cancer Working Group 3; PRO, patient-reported outcomes; PRO CoMiDa, patient-reported outcome completion and missing data; RECIST, Response Evaluation Criteria in Solid Tumours; QLQ, quality of life questionnaire; Q3C, every third cycle; WBBS, whole-body bone scan
One cycle is 21 days; in exceptional circumstances, the scheduled day of treatment (day 1) should be in accordance with local hospital practices.
All patients will be followed up until the last patient completes 2 years of follow-up.
Circulating mGSTP1: In pre-screening to be assessed within 28 days prior to day 1 cycle 1 and within 3 days prior to day 1 dosing in cycle 3. For patients in arm A in the main study, circulating mGSTP1 testing to be performed every third cycle after the pre-day 1 cycle 3 test until either unacceptable toxicity or disease progression.
Physical examination: At screening for the main study, a full physical examination is required.
Vital signs, including height and weight: respiratory rate, heart rate, blood pressure, O2 saturation (by pulse oximeter), and temperature. Height only required at screening for the main study.
Haematology: White blood cell (WCC), neutrophils/ANC, lymphocytes, platelets, haemoglobin. In pre-screening, to be performed within 14 days prior to cycle 1 day 1. In screening for the main study, to be performed within 21 days prior to randomisation.
Biochemistry: In pre-screening bilirubin, alkaline phosphatase (ALP), AST, ALT, and LDH, to be performed within 14 days prior to cycle 1 day 1. In screening for the main study and End of Treatment visit, ALP and LDH are required, to be performed within 21 days prior to randomisation.
PSA: In screening for the main study, to be performed within 21 days prior to randomisation.
CT (chest/abdomen/pelvis)/MRI scan: To be performed within 28 days prior to cycle 1 day 1 in the pre-screening phase, then every 12 weeks ± 5 days after cycle 1 day 1 during the treatment phase. In the follow-up phase, a CT scan is to be performed every 12 weeks ± 5 days after cycle 1 day 1 for the first 12 months and then every 24 weeks ± 5 days thereafter until disease progression according to RECIST 1.1 or PCWG3.
Whole-body bone scan: To be performed within 28 days prior to cycle 1 day 1 in the pre-screening phase, then every 12 weeks ± 5 days after cycle 1 day 1 during the treatment phase. In the follow-up phase, a whole-body bone scan is to be performed every 12 weeks ± 5 days after cycle 1 day 1 for the first 12 months and then every 24 weeks ± 5 days thereafter until disease progression according to RECIST 1.1 or PCWG3.
Patient-reported outcomes: To be performed within 5 days of randomisation and then every 12 weeks ± 5 days from cycle 1 day 1 until disease progression according to RECIST1.1 or PCWG3 criteria. (PRO completion to be timed with CT scans).
Patients in Arm A will stop docetaxel until mGSTP1 is detectable (and/or PSA rise >25% over nadir or clinicians’ discretion). During treatment holidays, all treatment phase assessments must be continued. Patients in Arm B will continue docetaxel in accordance with clinicians’ discretion or usual practice.
PROs
PRO data will be collected from patients participating in GUIDE during the treatment and follow-up phases to explore the impacts that intermittent and standard of care docetaxel have on quality of life. Four commonly used instruments will be used in GUIDE to evaluate PRO. The European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Cancer 30 version 3 (QLQ-C30) is a comprehensive and reliable tool with discriminatory validity evaluating functional domains and symptoms in people with cancer. The EORTC Fatigue scale (FA-12) will assess physical, cognitive, and emotional aspects of cancer-related fatigue. The Short Fear of Progression Questionnaire (FoP-Q-SF) evaluates peoples’ fear of cancer progression and has demonstrated reliability and validity. Finally, the modified Patient-Report Outcomes Common Terminology Criteria for Adverse Events (PRO-CTCAE) explores symptom burden and treatment toxicity as reported by patients.
The ANZUP Consumer Advisory Panel (CAP), which comprises a group of engaged consumer representatives, was involved in the selection of PRO instruments and the development of the GUIDE protocol and participant recruitment materials, including the participant information and consent form.
Resource use
Resource use associated with mGSTP1 directed therapy will be measured from trial-based case report forms and will include frequency of mGSTP1 testing, use of docetaxel and corticosteroids, pathology tests and imaging. Participants participating in GUIDE will be consented for access to their Medicare data, Australia’s administrative claims database which will provide information on their outpatient use of publicly subsidised drug therapies and medical services (including clinical, diagnostic, and imaging services).
Statistical considerations
The primary endpoint for GUIDE is rPFS, defined as the time from randomisation to the date of first documented progression on imaging by site investigator (PCWG3 criteria for bone lesions and RECIST v1.1 for soft tissue lesions), or death due to any cause in the whole population.
Based on the post hoc analysis of the SYNERGY trial, we expect the 12-month rPFS to be 50%. We assumed that an rPFS curve with a 35% 12-month rPFS is not desirable (H0) for people treated with intermittent docetaxel. Using a one-sided log-rank test with 2.5% alpha and 90% power, the number of evaluable participants treated with intermittent docetaxel is 69 assuming exponential rPFS distribution, and that recruitment will occur uniformly over 3 years and participants will be followed until the last patient has 2 years of follow-up. To allow for up to 10% of dropout prior to radiographic progression, at least 77 participants are required to be treated with intermittent docetaxel. Therefore, 120 people with undetectable mGSPT1 after two cycles of docetaxel will be randomised at 2:1 ratio between the arms. The undetectable mGSPT1 rate after two cycles is expected to be approximately 50%; therefore, 240 people with detectable mGSPT1 at baseline will be required in pre-screening. At baseline, the detectable mGSTP1 rate is around 80%, hence 300 people will be required to undergo pre-screening.
Analysis plan
Baseline characteristics and treatment details will be described using descriptive statistics such as minimum, maximum, median, interquartile range, mean and standard deviation for quantitative variables. Categorical variables will be described in tabular form as counts and percentages.
Point estimates for rPFS and OS will be estimated using the Kaplan–Meier method with 95% CIs at key time points (e.g. 12 months). Median times will also be provided with 95% CIs if they can be estimated. Kaplan–Meier curves will be produced to summarise the distribution of the time-to-event data. HRs and 95% CIs will be estimated using the stratified Cox model using prior novel anti-androgen therapy as stratification factor.
The maximum toxicity grade per participant of each AE will be derived and presented in table format. The analysis of AEs will be conducted twice: once for all AEs, regardless of relatedness to treatment and once considering only AEs related to the treatment. Listing of all SAE will be provided.
PRO will be analysed using linear mixed models (LMMs) with treatment arm, time (as factor), and interaction between treatment arm and time included as a fixed effect, and participant included as a random effect. No within-group correlations will be assumed, with the model being fitted by maximising the restricted log-likelihood. Means and 95% CIs will be estimated from the LMM contrasts for each time point and each arm. PRO trajectory will be described graphically. Publicly available fees and prices will be applied to items of resource use to estimate costs. Mean costs per treatment group (Arms A and B) will be reported and CIs generated by boot-strapping the data.
For the intermittent population, the time on treatment holiday will be described using the Kaplan–Meier method. Participants still on treatment at the end of the study will be censored. A Kaplan–Meier curve will be produced and the median time on treatment holiday will be estimated with 95% CI. The number of treatment holidays will be tabulated as count and percentages. A rate per year will also be provided with number of treatment holidays in the numerator and treatment duration in the denominator.
The proportion of participants who could have had a treatment holiday based on PSA will be described as a rate with 95% CI.
Results
The study endpoints are summarised in Table 3. The study was opened to enrolment November 2021.
Study endpoints.

Discussion
Docetaxel has a clear role in the management of metastatic CRPC; however, its toxicity is cumulative and frequently prohibitive in an older population. De-escalation strategies are attractive and have been a focus of clinical interest for several years. These efforts have been limited by an absence of suitable biomarkers of response to guide treatment.
The biomarker development process from discovery and assay development through to clinical practice is complex. 26 In brief, a biomarker needs to successfully progress through three key phases of testing which may involve multiple studies at each phase. The three phases are analytical validity, clinical validity, and clinical utility. 27 Analytical validity is a technical assessment to ensure the biomarker assay can deliver reproducible, accurate results using the appropriate patient specimen. Clinical validity assesses whether a biomarker can separate a patient population into two or more groups with distinct clinical or biological outcomes. The final crucial step, clinical utility, is designed to test whether a biomarker used to guide clinical practice actually improves patient outcomes compared to when that biomarker is not utilised. 26 While the literature is inundated with biomarkers with potential prognostic and/or predictive value, few reach testing beyond clinical validity. Although many biomarkers have an available commercial assay, only a minority achieve the required validation to be entered into clinical practice guidelines and applied to patient care. 28
The relevance of mGSTP1 has evolved in the last decade with growing evidence demonstrating its analytical and clinical validity as a novel epigenetic ctDNA biomarker. In an exploratory cohort of men with metastatic CRPC, mGSTP1 ctDNA was reliably assayed with good concordance in plasma and serum and was more sensitive than older, more cumbersome methods using circulating tumour cells. 13 Detectable mGSTP1 was then defined as >1 ng/mL of plasma utilising this method. This study established the analytical validity of the mGSTP1 ctDNA assay when applied to plasma and serum samples.
Clinical validity was then tested, initially in an independent, internal validation cohort of men with mCRPC receiving chemotherapy showing that mGSTP1 ctDNA was strongly associated with prognosis at baseline and after one cycle of docetaxel chemotherapy. 16 Further evidence was established in a post hoc analysis of samples from a large phase-3 trial, SYNERGY. 29 The SYNERGY study was a large, international, randomised controlled trial of men with mCRPC receiving first-line systemic therapy with docetaxel and custirsen or placebo. No benefit was found for the addition of custirsen. Serum samples from 600 patients taken at baseline and just prior to cycle 3 were assayed for mGSTP1 ctDNA. A fall in mGSTP1 ctDNA to undetectable levels after two cycles of docetaxel chemotherapy clearly conferred a favourable prognosis (HR for OS 0.36, 95% CI 0.29–0.46; p < 0.00001). 17 Level-II evidence of clinical validity was established. 30
While the evidence to date for mGSTP1 ctDNA has relied on testing of archived specimens, the GUIDE study has been specifically designed to test the use of this biomarker in guiding clinical decisions. If successful, the GUIDE study will be a crucial step towards proving clinical utility for mGSTP1 ctDNA and advancing this biomarker into the clinic to the benefit of our patients.
A non-comparative phase-2 study design was chosen to provide a contemporary and prospective standard of care arm. This is important to verify our null hypothesis and confirm our estimated rPFS curves are accurate. If GUIDE meets its primary endpoint, a randomised controlled phase-3 trial will be developed. The primary endpoint, rPFS was chosen to allow early analysis of the clinical utility of this novel approach as we anticipate that the 12-month rPFS rate to be approximately 50%. Second, rPFS has now been shown to be a robust end point in multiple trials with reasonable correlation with OS,31,32 making it highly clinically relevant. Of the secondary endpoints, we are particularly interested to document duration of treatment holidays and PROs. Given the concerns regarding tolerability of docetaxel in older men with metastatic CRPC, if GUIDE can offer safe treatment de-escalation, this may in turn improve tolerability of treatment and importantly optimise the quality of life for this group of men.
Conclusion
GUIDE will provide the first prospective evidence regarding the clinical utility of plasma mGSTP1 ctDNA, building upon existing data supporting analytical and clinical validity. We anticipate that this novel biomarker will safely guide treatment de-escalation and improve the quality of survival for a group of men with metastatic CRPC.
Footnotes
Author contributions
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ANZUP, Chris O’Brien Lifehouse Philanthropic Fund and the Twin Towns Foundation, CINSW TPG (TPG172146), and NHMRC investigator grant (APP1196225).
Collaborators and study organisation
The study is a locally developed and investigator-initiated academic trial sponsored by the Australian and New Zealand Urogenital and Prostate Trials Group (ANZUP). The trial is being coordinated by the Centre of Biostatistics and Clinical Trials (BaCT).
The trial management committee oversees study planning, monitoring, progress and review or information from related research.
Participating centres
The following centres are participating (principal investigators in brackets): Chris O’Brien Lifehouse (Kate Mahon), Concord Repatriation General Hospital (Anthony Linton), The Kinghorn Cancer Centre (Megan Crumbaker), Dubbo Base Hospital (Florian Honeyball), Albury Wodonga Regional Cancer Centre (Craig Underhill), and Goulburn Valley Health (Javier Torres).