
Abstract
Background:
Estrogen receptor positive (ER+) breast cancer is one of the most commonly diagnosed malignancies in women irrespective of their race or ethnicity. While Black women with ER+ breast cancer are 42% more likely to die of their disease than White women, molecular mechanisms underlying this disparate outcome are understudied. Recent studies identify DNA damage repair (DDR) genes as a new class of endocrine therapy resistance driver that contributes to poor survival among ER+ breast cancer patients. Here, we systematically analyze DDR regulation in the tumors and normal breast of Black women and its impact on survival outcome.
Method:
Mutation and up/downregulation of 104 DDR genes in breast tumor and normal samples from Black patients relative to White counterparts was assessed. For DDR genes that were differently regulated in the tumor samples from Black women in multiple datasets associations with survival outcome were tested.
Results:
Overall, Black patient tumors upregulate or downregulate RNA levels of a wide array of single strand break repair (SSBR) genes relative to their white counterparts and uniformly upregulate double strand break repair (DSBR) genes. This DSBR upregulation was also detectable in samples of normal breast tissue from Black women. Eight candidate DDR genes were reproducibly differently regulated in tumors from Black women and associated with poor survival. A unique DDR signature comprised of simultaneous upregulation of homologous recombination gene expression and downregulation of SSBR genes was enriched in Black patients. This signature associated with cell cycle dysregulation (p < 0.001), a hallmark of endocrine therapy resistance, and concordantly, with significantly worse survival outcomes in all datasets analyzed (hazard ratio of 9.5, p < 0.001).
Conclusion:
These results constitute the first systematic analysis of DDR regulation in Black women and provide strong rationale for refining biomarker profiles to ensure precision medicine for underserved populations.
Keywords
Introduction
Black people have the highest mortality rate across cancer types. 1 As in White women, the three most common cancer diagnoses in Black women are breast, lung, and colorectal cancer.1–4 Breast cancer accounts for 32% of these diagnoses, making it one of the most predominant cancer types in Black women. 4 Although Black women typically have higher incidence of triple negative breast cancer than white women, estrogen receptor positive (ER+) breast cancer remains the most commonly diagnosed subtype of breast cancer in Black women.2–4 Compared with white women with ER+ breast cancer, Black women are more likely to present with high grade and luminal B disease, which is less responsive to standard-of-care endocrine therapy. 5 In accordance with this difference in presentation, Black patients with ER+ breast cancer have a 42% higher mortality rate than White patients.6–10
Environmental, lifestyle, and socioeconomic factors, including access to healthcare,11–16 contribute to poor breast cancer outcomes in Black women.6,7,17,18 However, even after these factors are controlled for, differences in presentation and outcome persist in Black patients with ER+ breast cancer. 14 Therefore, it seems likely that ER+ breast cancer in Black patients has a distinct molecular journey19–21 that has to be better understood if precision medicine approaches are to be tailored to this underserved population. Such efforts require comprehensive characterization of tumors and normal tissue from Black women.
The severe underrepresentation of Black women in currently available datasets of patient tumors, however, presents a formidable challenge to agnostic and comprehensive characterization of unique molecular drivers and mechanisms. This is true even of ER+ breast cancer, which is one of the most researched cancer types with large and multiple datasets comprised of whole genome/exome sequencing and gene expression from thousands of patient tumors. Although these datasets have too few tumors from Black women to power agnostic screens, it is possible to conduct exploratory hypothesis-based analyses to provide proof-of-concept that tumors from Black women have distinct molecular profiles.
We, and others, have previously shown a link between DNA damage repair (DDR) dysregulation and endocrine therapy resistance in ER+ breast cancer.22–24 Specifically, we identified causal links between defects in the MutL complex of mismatch repair (MMR), CETN2 and ERCC1 from nucleotide excision repair (NER) and NEIL2 from base excision repair (BER) to cell cycle dysregulation and thereby, resistance to endocrine therapies in ER+ breast cancer. 24 However, our studies generated molecular hypotheses from datasets where Black patients were unrepresented (Figure S1A–B). Of the datasets used in our original analysis, only TCGA included data from Black women (n = 49 ER+ tumors from Black women versus n = 301 tumors from White women). 24 Therefore, results from this original analysis only reflect the DDR landscape of ER+ tumors, and its association with outcome, for white women. Since outcomes are worse for Black compared to white patients with ER+ breast cancer,2,11,25 here we test the hypothesis that higher frequency of dysregulation of DDR genes leading to resistance to endocrine therapy in tumors from Black women contributes to this outcome disparity.
Materials and methods
DNA damage repair gene set compilation
Gene sets for all DDR pathways analyzed (MMR, NER, BER, nonhomologous end joining (NHEJ), Fanconi anemia (FA), and homologous recombination (HR)) were derived from our previously published, curated DDR gene list. 24 Genes shared across different DDR pathways and downstream DDR checkpoint genes were not included as they complicate the evaluation of the relative contribution of individual DDR pathways to the cell cycle phenotypes and survival outcomes assessed in this study.
Datasets
Tumor datasets
The first dataset (GEO78958, referred to henceforth as dataset #958) has gene expression microarray data from tumors from 51 Black women with luminal (ER+) breast cancer, and 169 White women. 11 A second dataset (GSE18229, referred to henceforth as dataset #229) has gene expression microarray data from tumors from 44 Black and 85 White women. 26 This dataset includes tumors of all subtypes although ~70% of tumors are ER+ /luminal. A third dataset is the subset of ER+ tumors (irrespective of HER2 status) from TCGA and consists of RNAseq gene expression and whole exome sequence data from 49 tumors from Black and 449 from White women. 27 TCGA mutation data (downloaded March 2020) were obtained from cBioPortal. Gene expression from dataset #958 were available through the Gene Expression Omnibus (GEO, GSE78958), for dataset #229 were downloaded from dbGaP (downloaded May 2020) and for TCGA (downloaded March 2020) were obtained from cBioPortal. TCGA survival outcomes were downloaded from cBioPortal 27 (downloaded May 2020). Standard cutoffs of mean ± 1.5× standard deviation (SD) were used on the RNA data to identify ‘High’ and ‘Low’ subsets, respectively, for DDR genes in each dataset. Cut-offs using 0.5 SD were used to categorize CDK genes as ‘High’ and ‘Low’. Details of the datasets are presented in Tables S1–S3. All tumor data originated from primary samples. Tumors in both TCGA and the Ellsworth dataset (GEO78958) were treatment-naïve. The treatment status of tumors from the third dataset (GSE18229) is uncertain.
Normal datasets
A normal breast tissue dataset (GSE43973), 28 has gene expression microarray data from 12 Black and 98 white women. A second tumor adjacent normal tissue dataset (GSE50939) 29 has gene expression microarray data from 14 Black and 52 white patients. Both these datasets were downloaded from GEO (March 2020). 24
Enrichment analysis
For RNA analysis, p values were obtained by comparing each gene between tumors from Black and white women using the two-tailed Wilcoxon Rank Sum test within each dataset analyzed. For each dataset, the false discovery rate (FDR) was calculated and genes with an FDR ⩽0.2 were prioritized. 30 Genes that made the FDR cut-off in at least two out of three datasets were considered candidates for subsequent hypothesis-based analyses. Two-tailed Fisher’s exact test determined p values for overall patterns of upregulation and downregulation. The p values comparing overall patterns of regulation were adjusted for multiple comparisons using the Holm’s method. 31
For mutation analysis, any DDR gene which harbored >1 somatic mutation in tumors from Black women in TCGA was considered. All protein changing mutations were included irrespective of category (i.e. missense, nonsense, frameshift) or in silico predicted pathogenicity. Expected rates of mutation frequency were calculated based on the total number of mutations identified in the entire patient population and compared to observed rates in tumors from Black and white patients respectively. Two-tailed Fisher’s exact test determined p values by comparing observed to expected frequency of mutations in each gene.
For functional analysis, the functional category was determined based on literature searches for each gene of each pathway used in this targeted analysis (detailed in Table S4). Each gene was then assigned to one or more of three categories: Sensor, Scaffold, and Repair. ‘Sensors’ sense the presence of specific types of DNA damage, ‘Scaffold’ proteins serve to stabilize and activate cell cycle checkpoint kinases, ATM/ATR, at the site of damaged DNA, while ‘Repair’ proteins are directly involved in repairing damaged DNA. If a gene fell into two functional categories, it was considered in statistical analyses of each category in turn. The number of candidate genes in each category was compared to the total number of genes in that category using a two-tailed Fisher’s exact test.
For global pathway analysis from two normal breast datasets, two-tailed Wilcoxon Rank Sum p-values were used to prioritize genes in Black versus White women in each dataset and all prioritized genes appearing at least once in either dataset analyzed were included in statistical analyses. For proliferation analyses, Ki67 RNA levels (gene name: MKI67) were used and the top 20% and bottom 20% of Ki67 expressing tumors were considered as ‘High’ and ‘Low’ proliferators respectively. For age analysis, menopausal status was used as a surrogate. Fisher’s exact test determined p values for categorical analyses.
Survival analysis
For Kaplan–Meier analyses, all tumors with associated survival data in each dataset were used, with restriction to luminal A/B tumors in dataset #958 and ER+ tumors in TCGA. Outcome measures used were disease-free survival for dataset #958, recurrence-free survival in dataset #229 and overall survival for TCGA. Outcome measures were selected for each dataset based on having the largest sample size. Only samples with survival metadata were included in the analysis. Log rank test calculated p values.
Results
Landscape of DNA repair regulation in ER+ breast tumors from Black women
We first assessed the expression of DDR genes previously associated with poor survival in white women (MLH1, PMS2, CETN2, NEIL2, and ERCC1) 24 in ER+ tumors from Black women. There was no detectable enrichment in frequency of downregulation of these genes in tumors from Black women relative to white women (Figure S1C). Furthermore, we found no increase in disease recurrence when comparing tumors with downregulation of these DDR genes versus those without in Black women, in contrast to the observed, and expected, increase in recurrence in white women (Figure S1D). Therefore, to investigate whether ER+ tumors from Black patients have a distinct pattern of DDR regulation that contributes to poor outcomes, we assessed RNA levels of 104 DDR genes from six principal DDR pathways in each of three datasets described in Tables S1–3 (Figure 1).

Schematic of the study outline. Three tumor and two normal breast datasets were analyzed for differences in RNA levels of 104 DNA damage repair (DDR) genes from six pathways to map the landscape of DDR in Black (B) versus White (W) women. DDR genes differently expressed in tumors from Black women in ⩾2 tumor datasets were assessed in survival analyses in all three datasets. In parallel, differences in RNA levels of DDR genes between Black and White women were interrogated at pathway level in two normal breast datasets.
Sixty-seven DDR genes were either upregulated or downregulated in tumors from Black patients relative to those from white patients in at least one of the three tumor datasets analyzed (Figure S2). Single strand break repair (SSBR) genes, specifically genes from NER and BER pathways were enriched in this differently expressed gene set (Figure 2(a)). Overall, genes from double strand break repair (DSBR) pathways (Fanconi anemia: FA and homologous recombination: HR) were predominantly upregulated in tumors from Black patients (Figure 2(b), Figure S2). Eight DDR genes were significantly up/downregulated in at least two of the three datasets analyzed. Three of these eight genes were downregulated (two BER genes: XRCC1, PARP1 and one nonhomologous end joining (NHEJ) gene: XRCC4), and the other five were upregulated (BER: NEIL3, NER: MNAT1, FA: FANCE and HR: NBN and BRCA1) in tumors from Black women when compared to those from white women (Figure 2(c)).

Tumors from Black breast cancer patients have a distinct DNA repair landscape. (a) Venn diagram showing proportion of genes from each of six DDR pathways that are significantly dysregulated in tumors from Black (B) women relative to those from white (W) women in at least one of three datasets analyzed. Associated Figure S2 presents full list of DDR genes included here. (b) Stacked column graphs representing number of DDR genes that are up- (yellow) or down (blue)-regulated in tumors from Black versus White patients by pathway. Pearson’s chi-square test determined p values. (c) Heatmap showing candidate genes that are either upregulated (yellow) or downregulated (blue) in at least two datasets as assessed by q-value analysis based on p values derived from two-tailed Wilcoxon Rank Sum tests. (d) Bar graph showing observed/expected ratio for mutational frequency of all DDR genes mutated at least once in ER+ tumors from Black women in TCGA. Observed/expected ratio for tumors from white patients (pts) are represented by bars to the left of the median line, and for tumors from Black patients to the right. Fisher’s Exact test determined p values. Figure S3 presents associated data on DDR gene mutations in tumors from Black and white breast cancer patients.
Using whole exome sequence data from TCGA, we found that of all eight differently expressed DDR genes, only PARP1 (BER; p = 0.01) and BRCA1 (HR; p = 0.03) were significantly enriched for mutation in tumors from Black patients (Figure S3A). In an agnostic analysis across all 104 DDR genes, 16% of tumors from Black patients had mutations in at least one DDR gene, compared to only 3% of tumors from white patients (p < 0.001), with specific enrichment for mutations in genes from BER and HR pathways (Figure S3B). In addition to PARP1 and BRCA1, ERCC6 (NER), PARP4 (BER), FANCM and FAAP24 (FA) also had increased mutational frequency in tumors from Black versus White women (Figure 2(d)). Mutations in any DDR gene associated with significantly worse disease-free survival in Black patients (Hazard Ratio = 4.12, p = 0.02, Figure S3 C), supporting a clinically relevant role for the mutation of specific DDR genes in the poor outcomes observed in this patient cohort.
Functional pattern associated with DNA repair landscape in tumors from Black women
Next, we assessed the distribution of the 8 shortlisted DDR genes that were differently expressed in tumors from Black patients into functional categories based on their primary function as Sensor, cell cycle checkpoint (ATM/ATR) Scaffold or Repair (see Table S4). Components of all DDR pathways predominantly function as Sensor or Repair proteins, with only 15% of proteins in any DDR pathway constituting a Scaffold (Figure 3(a)). However, ~60% of DDR genes differently expressed in tumors from Black breast cancer patients are ATM/ATR Scaffolds (Figure 3(a)), resulting in a significant enrichment in the candidate DDR gene list for association with cell cycle regulation (p = 0.004). ATM/ATR are the key regulators of cell cycle response to DNA strand breaks and replication stress. 32 ATM plays a crucial role in the activation of the G1 cell cycle checkpoint, which prevents cells with damaged DNA from entering the S-phase. ATR plays an important role in regulating the intra-S and G2/M cell cycle checkpoints. The activation of these cell cycle checkpoints through DNA damage signaling inhibits specific cyclin-dependent kinases (CDKs), which are crucial for cell cycle progression. 33

Cell cycle dysregulation patterns associated with the DNA repair landscape of ER+ tumors from Black patients. (a) Representation of functional DNA repair categories in 104 DDR genes screened (All) and in the list of eight genes (Candidates) identified in Figure 2(c). Fisher’s exact test determined p values. ATM/Chk2 and ATR/Chk1 scaffolds were combined to represent cell cycle checkpoint scaffolds in statistical analyses. (b) Heat map demonstrating up-(yellow) or down-(blue)-regulation of the four principle CDKs in tumors from #958 and TCGA with dysregulation of the eight shortlisted DDR genes, grouped based on pathway as indicated. The third dataset was not included in this analysis as it is missing RNA data from all CDK genes. Non-HR and non-BER genes were grouped as Other DDR. Asterisks indicate statistical significance (p < 0.05*, p < 0.01**, p < 0.001***) from two-tailed Wilcoxon Rank Sum tests after Holm’s adjustment for multiple comparisons) and outlined boxes indicate genes with consistent statistical significance across datasets. Schematic of the cell cycle included for context. (c) PCNA RNA analysis in TCGA. Multivariate ANOVA test with pairwise comparison determined p value. Supporting data in Figure S4.
To test whether differential regulation of the eight shortlisted DDR genes associates with cell cycle dysregulation, we next analyzed RNA levels of each of four principal CDKs: CDK1 (G2/M), CDK2 (S), and CDK4/6 (G1) in tumors with upregulation or downregulation of the eight shortlisted DDR genes. Since a high proportion of tumors demonstrated differential expression of HR or BER genes, we were able to assess CDK RNA levels specifically in relation to these pathways. A subset of tumors had coincident upregulation and downregulation of HR and BER genes, respectively, referred to as HR/BER tumors. Tumors with differential expression of genes from any of the other DDR pathways were grouped together as their numbers were insufficient to allow independent analysis of each pathway. The HR/BER subset significantly upregulated CDK2, a positive regulator of the S phase of the cell cycle, relative to all other subsets across datasets (Figure 3(b)). In addition, in TCGA, HR/BER tumors significantly upregulated PCNA, another marker of S phase progression (Figure 3(c)). This increase in S phase cell cycle markers is in accordance with increased overall proliferation levels in ER+ tumors from Black women relative to those from white women (Figure S4). When comparing CDK gene expression between Black and White patient tumors independent of DDR gene expression, we found significant upregulation of CDK1, CDK4, and CDK6 in tumors from Black patients (p = 8.05e−05, p = 0.0001, and p = 0.004, respectively), but no difference in CDK2. These data suggest a new association between S phase progression and the DDR landscape of ER+ tumors from Black women.
Normal breast tissue from Black women has high expression of double strand break repair genes relative to normal breast tissue from white women
We next analyzed gene expression microarray data from two publicly available datasets of noncancer breast tissue: GSE43973 with samples from reduction mammoplasty (12 Black and 98 White women, henceforth termed normal) and GSE50939 with samples from tumor-adjacent normal tissue (14 Black and 67 White breast cancer patients, henceforth termed adjacent).28,29 Overall, the predominant difference in DDR gene expression between breast tissue from Black and White women is higher expression of DSBR genes (Figure 4(a)–(c)).

Differences in the DNA repair landscape of the normal breast in Black versus White women. (a) Nested donut plots representing the proportion of SSBR (black) versus DSBR (gray) genes (outer donut) and proportion of genes from each pathway within SSBR and DSBR categories (inner donut) that were significantly (two-tailed Wilcoxon Rank Sum test) upregulated or downregulated in Black versus White normal breast samples. Fisher’s Exact test determined p values. (b and c) Stacked column graphs summarizing numbers of DSBR (b) and SSBR (c) genes that were either upregulated (yellow) or down-(blue)-regulated in normal (Normal and Adj) versus tumor (#958, #229, TCGA) datasets. Supporting data in Figure S5. (d) Stacked column graphs representing the proportion of high and low proliferating samples (left) and pre- and post-menopausal women (right) in normal datasets. High proliferating samples reflect the top 20% of samples ranked by gene expression of Ki67 (MKI67, gene name), a marker of proliferation from high to low, while low proliferating samples reflect the bottom 20th percentile. Fisher’s Exact test determined all categorical p values.
Even NHEJ genes, which are preferentially downregulated in tumors from Black women, are upregulated in the normal breast of Black women when compared to that of White women (Figure 4(b), Figure S5). This is a distinct pattern in comparison to tumors where the majority of differently expressed DDR genes are from SSBR pathways (Figure 4(a) and (c)). Differential expression of MMR genes appears restricted to ER+ tumors from Black women with no detectable differences in the normal breast (Figure 4(a) and S5). In contrast, FA genes are frequently differently expressed in the normal breast of Black women relative to that of White women, but less so in their tumors (Figure 4(a) and S5). The relative impact of this differential DDR gene expression in the normal breast tissue of Black women on response to therapy or survival outcomes remains to be investigated.
Both proliferation and age are known to affect DDR gene expression. 28 However, in combined data from normal and tumor-adjacent normal datasets, using gene expression of MKI67 as a proliferative index, we found no difference in baseline proliferation of normal breast tissue between Black and White women (45% versus 49% were highly proliferative, Figure 4(d)). Similarly, the menopausal status of Black versus White women, a surrogate marker for age, in the normal dataset is entirely comparable (Figure 4(d)). Overall, these data identify endogenous differences in the DDR landscape of the normal breast in Black versus White women, and neither baseline proliferation nor age explain these differences.
The DNA repair landscape of ER+ breast tumors from Black women associates with poor outcome
To assess the clinical relevance of the DNA repair landscape in Black women, we next determined the frequency of dysregulation of the eight DDR genes shortlisted from the analysis of ER+ tumors (Figure 2(c)) and association with survival outcome. Overall, 45–60% of ER+ tumors from Black women had detectable dysregulation of at least one of these eight DDR genes, compared to 25–30% of tumors from white women, a statistically significant difference in every dataset analyzed (Figure 5(a) and (b), Figure S6A). Notably, we observed consistent enrichment for coincident upregulation of one of two HR genes, NBN or BRCA1, with upregulation or downregulation of SSBR genes (an HR/SSBR subset) in tumors from Black women (6–8% versus 1–3% in tumors from White women; Figure 5(a) and (b)). Of note, this subset (HR/SSBR) is rare in TCGA (Figure S6A) likely because of the difference in gene expression platforms – TCGA data are obtained from RNAseq and overrepresents FA genes within the DSBR group relative to the other two gene expression microarray datasets analyzed (see Figure S2).
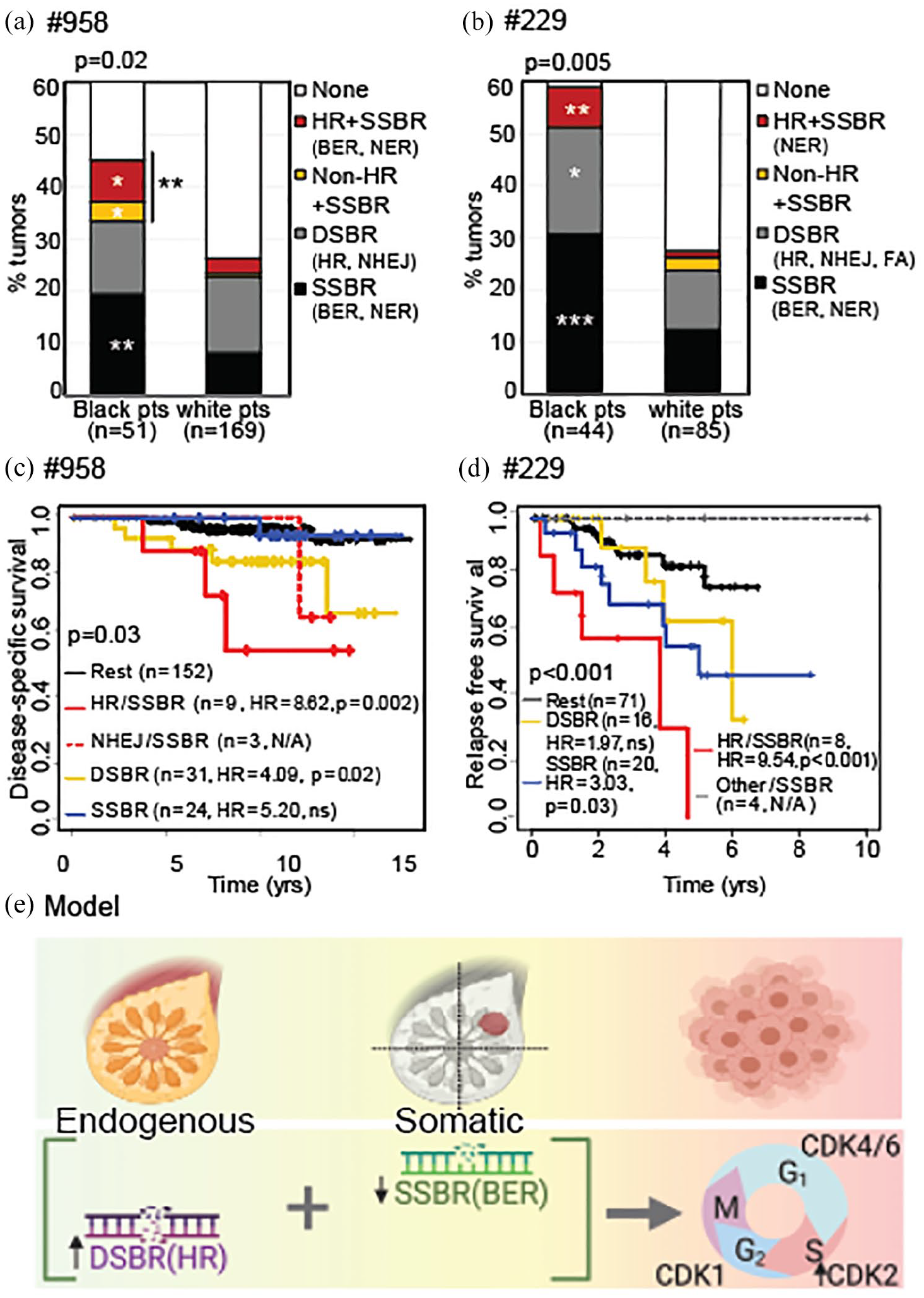
The DNA repair landscape of ER+ tumors from Black women associates with worse survival. (a and b) Stacked columns representing % of tumors from Black versus White patients (pts) with differential expression of any one of the eight shortlisted DDR genes from the specified pathways in #958 (a) and #229 (b). Pearson’s chi-square test determined p values. Similar analysis in TCGA in Figure S6A. (c and d) Kaplan–Meier curves representing disease-specific survival in #958 (c) and relapse-free survival in #229 (d) of patients whose tumors had differential expression of specified DDR genes, by pathway, relative to tumors that did not. Log rank test determined p values. HR in survival curves, hazard ratio, p < 0.05*; p < 0.01**; p < 0.001***. Associated data for TCGA presented in Figure S6B. (e) Working model proposing co-incidence of DSBR upregulation (primarily, of HR genes) in normal breast and upregulation or downregulation of SSBR genes (primarily downregulation of BER genes) during tumor formation and progression, in ER+ tumors from Black women. This co-incident altered DDR regulation associates with increased CDK2 gene expression, and significantly worse survival outcome.
We also observed consistent and significant association of the HR/SSBR tumor subset with worse disease-specific survival (dataset #958, HR = 8.62, p = 0.002, Figure 5(c)), relapse-free survival (dataset #229, HR = 9.54, p < 0.001, Figure 5(d)) and overall survival (TCGA, Figure S6B). Together, these results identify specific patterns of DSBR upregulation in normal breast and SSBR upregulation or downregulation in ER+ tumor tissue from Black women that are distinct from that seen in White women. Moreover, the DNA repair landscape uncovered in tumors from Black women associates with increased CDK2 gene expression and worse outcome by every disease measure analyzed across three independent datasets (Figure 5(e)).
Discussion
DNA repair proteins are natural molecular conduits between external stimuli and cellular response. Exposure to genotoxins or hypoxia, for instance, can induce a cell to upregulate or downregulate its DDR signaling. 34 Not only do DDR proteins repair damaged DNA, they also activate cell cycle checkpoints and engage apoptotic pathways. 35 Therefore, DDR pathways make an attractive starting point for understanding how molecular factors that translate environmental stressors into cellular phenotypes may differ by race/ethnicity. The evidence provided by this study for the existence of a DDR expression pattern that is enriched in the normal breast and ER+ tumors in Black women relative to White women and that is prognostic of worse outcome has implications for transforming precision medicine from a ‘one size fits all’ approach to tailoring by ancestry and/or ethnicity-related molecular features.
A role for individual HR or BER dysregulation in tumors from Black women, primarily for triple negative breast cancer, has been previously indicated.36–38 Results presented here that highlight the existence of a coincident regulation pattern for these two pathways in ER+ tumors from Black women, potentially originating from differences in DNA repair gene expression in the normal breast, adds to the literature and provides new insight into prognosis and outcomes in this underserved population. The upregulation of gene expression from NHEJ genes in the normal breast of Black women is intriguing, and whether it plays a causal role in the initiation of more aggressive tumors, due to its tendency to error-prone repair is worthy of further investigation in better powered datasets representing the normal breast tissue of Black women. 39 This is especially true given that proliferation levels in the normal breast tissue of Black and white women are comparable, while tumors in Black women are more proliferative than those in white women, suggesting tumor-specific events that contribute to these differences. Of note, no MMR gene was consistently downregulated in Black tumors across our three datasets. Given the previous discovery of MMR gene downregulation as a key mediator of poor outcome in white ER+ breast cancer patients, 23 it is possible that this molecular driver is shared between Black and white patients, and is therefore not detectably further downregulated in, or unique to, tumors from Black patients. However, as in our earlier study of the DNA repair landscape in ER+ tumors with poor versus good outcome from White women, 24 genes from the NER and BER pathways appeared to be the most frequently differently expressed in ER + tumors from Black women.
An association with cell cycle regulation indicates a functional basis for the DDR landscape identified in ER+ tumors from Black women. Previous work suggests that cell cycle regulation, specifically at the G1/S transition, is instrumental for appropriate response to endocrine therapy, the standard-of-care for ER+ breast cancer patients.23,24,40,41 Indeed, use of CDK4/6 inhibitors to inhibit the G1/S cell cycle is the only therapeutic strategy shown to prolong survival in advanced, endocrine therapy resistant ER+ breast cancer patients.42–44 The work presented here, showing a statistically significant association between the HR/SSBR signature observed in ER+ tumors from Black women and upregulation of CDK2, the cyclin dependent kinase instrumental in S phase progression, presents the intriguing therapeutic hypothesis that at least a subset of Black ER+ breast cancer patients may benefit from earlier intervention with CDK inhibitors in combination with endocrine therapy. There is precedence in the literature for differential efficacy of CDK4/6 inhibitors in ER + breast cancer patients based on race, although no previous studies specifically investigate efficacy in Black ER+ breast cancer patients, likely due to the significant underrepresentation of Black women in most clinical trials.45–48
Overall, this study presents a roadmap for developing more effectively personalized biomarkers to predict and perhaps, prevent poor outcomes for underserved patient populations. The scope of this study is limited by the underrepresentation of Black patient tumors in existing public datasets, highlighting a critical need for more diverse and representative datasets. This limitation precluded more in-depth gene resolution analysis of mutational and expression data since each dataset used was only sufficiently powered to find differences with high effect size. This also limited the analyses to a subset of DDR genes that are unique to individual DDR pathways, omitting DDR and checkpoint genes that are shared between different pathways, which could nevertheless significantly impact the cell cycle phenotypes and survival outcomes assessed in this study. It is likely that bigger datasets of Black patient tumors will uncover more differences in DDR gene expression that may be prognostic or predictive. Conducting larger scale studies of both tumor and normal breast tissue, as well as causally testing molecular hypotheses raised here will constitute important future steps toward improving precision medicine approaches to cancer specifically for underserved populations.
Supplemental Material
sj-docx-1-tam-10.1177_17588359221075458 – Supplemental material for The DNA damage repair landscape in Black women with breast cancer
Supplemental material, sj-docx-1-tam-10.1177_17588359221075458 for The DNA damage repair landscape in Black women with breast cancer by Aloran Mazumder, Athena Jimenez, Rachel E. Ellsworth, Stephen J. Freedland, Sophia George, Matthew N. Bainbridge and Svasti Haricharan in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We acknowledge Drs Melissa Troester, Professor, University of North Carolina for sharing gene expression data and Meenakshi Anurag, Assistant Professor, Baylor College of Medicine for sharing DDR gene lists without compensation.
Author contributions
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Haricharan has a provisional patent (US 63/106777) for using DNA repair genes as a biomarker for patient outcome. Dr Freedland is a consultant to Pfizer, Janssen, Astra Zeneca, Merck, and Clovis. All other authors declare no conflicts of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Design and conduct of the study were funded by Department of Defense Breakthrough (W81XWH-18-1-0034) and Susan G. Komen Career Catalyst (CCR18548157) awards (to SH). Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the U.S. Army. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers P30CA030199 and CA229613 (to SH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Dr Haricharan had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Dr Mazumder and Ms Jimenez (Sanford Burnham Prebys) also accessed the data and conducted analyses.
Ethical approval and informed patient consent
Ethics approval was not sought for the present study because all data were public and de-identified. All patients provided informed consent in the original studies.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.