
Abstract
Background:
Standard intravenous (IV) paclitaxel is associated with hypersensitivity/toxicity. Alternative IV formulations have improved tolerability but still require frequent hospital visits and IV infusion. DHP107 is a novel oral formulation of paclitaxel that is approved in South Korea for the treatment of gastric cancer.
Methods:
This multicenter, phase II study using a Simon’s two-stage design investigated the efficacy and safety of DHP107 200 mg/m2 administered orally twice daily on days 1, 8, and 15 every 4 weeks for the first-line treatment of recurrent or metastatic HER2-negative breast cancer.
Results:
Thirty-six patients were enrolled and 31 were assessable for efficacy. Patient median age was 57 years (range = 34–81) and 11 (31%) had triple-negative disease. A median of seven cycles (range = 1–28) of DHP107 was administered. Objective response rate was 55% (17 patients), all partial responses, according to the investigator’s decision and independent central review (ICR), and 44% (4/9 patients) in those with triple-negative disease. Disease control rate (partial response and stable disease) was 74% (23 patients) according to the investigator’s decision and ICR. In the intention-to-treat (ITT) population of all enrolled participants, the objective response rate was 50% (18/36 patients). Median progression-free survival was 8.9 months [95% confidence interval [CI]: 5.2–12.3) and median time to treatment failure was 8.0 months (95% CI: 4.2–10.0). DHP107 had an acceptable toxicity profile. All patients experienced treatment-emergent adverse events; the most common adverse events were decreased neutrophil count (81% all grades and 78% grade ⩾ 3) followed by peripheral sensory neuropathy (61% all grades and 8% grade 3). However, there was no febrile neutropenia or sepsis.
Conclusion:
DHP107 showed promising efficacy and acceptable tolerability in this phase II study and is currently being investigated in the OPTIMAL phase III study (NCT03315364).
Trial registration:
This trial was registered with ClinicalTrials.gov identifier: NCT03315364.
Introduction
Female breast cancer is the second most commonly diagnosed cancer in the world after lung cancer, and the leading cause of cancer death. 1 In South Korea in 2017, breast cancer was the second most prevalent cancer after thyroid in women. 2 In recurrent or metastatic human epidermal growth factor receptor 2 (HER2)-negative breast cancer, prolongation of survival and improvement of quality of life (QoL) are important because treatment is not curative. 3 Therefore, the toxicity and tolerability of any treatment must be taken into consideration. 3 Paclitaxel is a preferred single agent for the treatment of patients with stage IV or recurrent metastatic breast cancer and triple-negative tumors and germline BRCA1/2 mutations in the National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines. 3 Paclitaxel can be administered weekly (80 mg/m2) or every 3 weeks (175 mg/m2) but weekly administration has been shown to be more beneficial than 3-weekly administration in terms of overall survival (OS) in patients with locally advanced or metastatic breast cancer. 4
The standard intravenous (IV) formulation of paclitaxel includes the non-ionic surfactant polyoxyl-35-castor oil (Cremophor EL or Kolliphor EL, BASF, Ludwigshafen, Germany), which is associated with hypersensitivity reactions and toxicity,5–8 requires an infusion of 3 hours or longer, and premedication to help prevent hypersensitivity reactions.6–8 In addition, Cremophor EL forms micelles within plasma that entrap the active drug, resulting in increased systemic exposure, decreased elimination, and reduced antitumor activity.9–11 To overcome these issues, other formulations, such as a nanoparticle albumin-bound IV formulation (nab-paclitaxel) 12 and polymeric micelle paclitaxel, 13 have been developed.
Although Cremophor EL-free formulations have improved the tolerability of IV paclitaxel, treatment still involves frequent hospital visits. Oral administration of paclitaxel would be an attractive alternative that could facilitate more patient-friendly treatment. 14 DHP107 (Liporaxel, Daehwa Pharmaceutical Co. Ltd., Seoul, Korea) is a novel oral formulation composed of lipid ingredients and paclitaxel that is systemically absorbed without the need for P-glycoprotein inhibitors or Cremophor EL. 15 Based on the results of the phase III DREAM study, which showed that DHP107 was non-inferior to 3-weekly paclitaxel in terms of progression-free survival (PFS), 16 DHP107 was approved in Korea in 2016 for the treatment of advanced, metastatic, or locally recurrent gastric cancer. The aim of the current study was to investigate the antitumor activity and tolerability of DHP107 in the first-line treatment of recurrent or metastatic breast cancer.
Patients and methods
Study design and treatment
This was a multicenter, open-label, single-arm, phase II study using a Simon’s optimal two-stage design to minimize any disadvantage to the patients’ treatment due to potential ineffectiveness of the study drug (ClinicalTrials.gov identifier NCT03315364). In stage 1, if two or more of nine patients had an objective response, the study could move to stage 2. In stage 2, if nine or more of 34 patients had an objective response, DHP107 was considered to be effective and able to proceed to the confirmatory phase III study. DHP107 200 mg/m2 was administered orally twice daily approximately 1 hour after breakfast and dinner with no premedication for the prevention of hypersensitivity on days 1, 8, and 15 of a 4-week cycle. Administration of DHP107 was continued until the occurrence of progressive disease. The study was approved by the relevant Institutional Review Boards: Institutional Review Board Asan Medical Center (2017-1216); Korea University Guro Hospital Institutional Review Board (2017GR0021); Seoul National University College of Medicine/Seoul National University Hospital Institutional Review Board (H-1709-121-889); Ajou University Hospital Institutional Review Board (AJIRB-MED-T23-17-334); Severance Hospital, Institutional Review Board (4-2017-0921); Chungbuk National University Hospital Institutional Review Board (CBNUH 2018-05-016); Korea University Anam Hospital Institutional Review Board (2017AN0346); CHA Bundang Medical Center Institutional Review Board (CHAMC 2018-05-040); Yonsei University, Wonju Severance Christian Hospital, Institutional Review Board (CR118024); Inje University Haeundae Paik Hospital Institutional Review Board (HPIRB 2018-05-022); and National Cancer Center Institutional Review Board (NCC2017-0261). It was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and local ethical and legal regulations. All patients provided written informed consent before enrollment.
Study end points and assessments
The primary end point was the investigator-assessed objective response rate (ORR). Secondary end points were the disease control rate (DCR), PFS, OS, time to treatment failure (TTF), QoL, and safety. Tumor response was evaluated using Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1) 17 every 8 weeks (±1 week) by the investigator and by independent central review (ICR) for the purpose of sensitivity analysis. ORR was assessed in the intention-to-treat (ITT) population of all randomized participants. QoL was assessed using the European Quality of Life (EuroQol) 5-Dimension 3-level (EQ-5D-3 L) instrument and the EQ visual analog scale (VAS).
Safety was assessed in all patients who received ⩾1 dose of allocated treatment. Safety was assessed throughout the study by monitoring the occurrence of treatment-emergent adverse events (TEAEs), laboratory tests, electrocardiogram, radiological examinations, left ventricular ejection fraction measurements, and Eastern Cooperative Oncology Group (ECOG) performance status. During treatment, patients underwent weekly blood counts, and a toxicity assessment was performed weekly for first the two cycles. Serum chemistries and a physical examination, with assessment of ECOG performance status and peripheral neuropathy, were performed before each cycle. The severity of TEAEs was determined according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
Patients
Male and female patients were eligible if they were ⩾19 years of age and had histologically or cytologically confirmed recurrent or metastatic HER2-negative breast cancer irrespective of hormone receptor (HR) status [estrogen receptor (ER) and progesterone receptor (PR) positive or negative]. Other inclusion criteria included measurable lesions, identified by revised RECIST (version 1.1), 16 ECOG performance status 0 or 1, and adequate hematologic [absolute neutrophil count (ANC) ⩾ 1500/mm3, platelet count ⩾ 100,000/mm3, hemoglobin ⩾ 9.0 g/dL], renal [serum creatinine ⩽ 1.5 × upper limit of normal (ULN), serum calcium level ⩽ 12 mg/dL], and liver function [total bilirubin ⩽ 1.5 × ULN, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ⩽ 2.5 × ULN or ⩽ 5 × ULN if documented liver metastases].
Key exclusion criteria were prior treatment with a taxane in the metastatic setting; prior chemotherapy for recurrent or metastatic HER2-negative breast cancer; the presence of cardiovascular disease or uncontrolled hypertension; pregnant or breast-feeding women. However, the following patients could participate if the last administration of adjuvant or neoadjuvant chemotherapy for breast cancer that was not recurrent or metastatic was ⩾1 year before randomization: ER+ or PR+ patients who had up to second-line endocrine therapy with or without concomitant cyclin-dependent kinase (CDK) 4/6 inhibitors.
Statistical analysis
Analyses were performed on the per-protocol set (PPS) and the safety analysis set (SAS). The PPS included patients without any major protocol violations who received at least one cycle of treatment and had at least one tumor assessment following the administration of DHP107. The SAS included all patients who received at least one dose of DHP107 and had at least one safety assessment. The PPS was used for the analysis of efficacy and the SAS for the analysis of demographics and safety.
Descriptive statistics are presented for continuous variables, and frequencies and percentages are presented for categorical variables. All end points include two-sided 95% confidence intervals (CIs). The number of patients was calculated by the Simon’s optimal two-stage design with a one-sided significance level of 0.05 and a test power of 80%. The ORR for paclitaxel as first-line treatment is 15–62% 18 and its weighted average is approximately 37%. Therefore, this study set the poor ORR (ineffectiveness) of DHP107 as 15% and the good ORR (effectiveness) was hypothesized to be 35%, which is close to the weighted average, to have a 20% difference between the poor and good ORR.
Results
Patients
Forty-five patients were screened and 36 were enrolled at eight sites in Korea between December 2017 and October 2018 (the trial was registered on 11 September 2017; Figure 1). Baseline demographic and clinical characteristics are shown in Table 1. The median age was 57 years (range = 34–81 years), and 11 patients (31%) had triple-negative disease. Common metastatic sites included bone (69%), lymph node (53%), and lung (50%). The majority of patients (67%) had received prior hormone therapy, with letrozole the most frequent prior adjuvant (42%) or neoadjuvant chemotherapy (81%), with an anthracycline-containing regimen the most frequent (72%).

CONSORT diagram.
Baseline patient demographic and clinical characteristics (N = 36).

ER, estrogen receptor; PR, progesterone receptor.
All patients received treatment with oral DHP107, and two patients remained on treatment at the data cutoff (30 January 2020). Thirty-four patients (94%) discontinued treatment: 23 (64%) due to disease progression, six (17%) due to adverse events (suicide attempt, n = 1; clear-cell carcinoma of right ovary, n = 1; oral mucositis, n = 1; peripheral sensory neuropathy, n = 3), two (6%) withdrew consent, and three (8%) for other reasons.
Efficacy
In the ITT population of all enrolled participants, the objective response rate was 50% (18/36 patients, Table 2). In the PPS analysis, in which five patients were excluded (three patients dropped out in the beginning and two patients were excluded due to prohibited medication), the ORR was 55% (17 patients), all of which were partial responses (PRs), according to both the investigator’s decision and ICR. The response rate was 44% (4/9 patients) in those with triple-negative disease (Figure 2). Six patients (19%) had stable disease (SD) by both the investigator’s decision and ICR. The DCR (PR+ SD) was 74% (23 patients) according to both the investigator’s decision and the ICR. Median PFS was 8.9 months (95% CI: 5.2–12.3) (Figure 3) and median TTF was 8.0 months (95% CI: 4.2–10.0) (Table 2) (Figure 4). OS was not reached for the survival probability of 0.5 so median OS is not estimable (NE) (95% CI: 18.1 months–NE) (Figure 5).
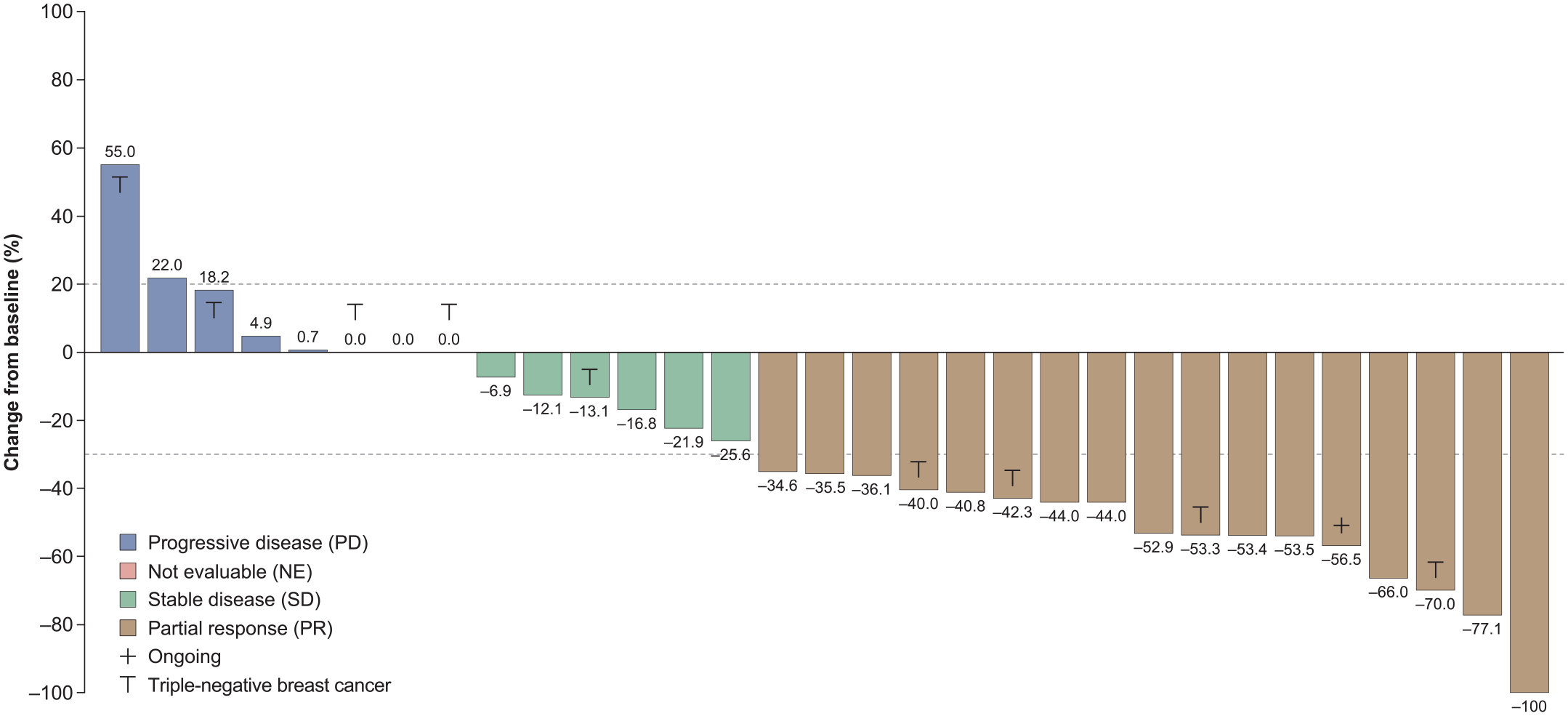
Change from baseline in tumor volume (per-protocol set).

Progression-free survival (per-protocol set).

Time to treatment failure (per-protocol set).

Overall survival (per-protocol set).
Response rate and survival in PP set and ITT set.
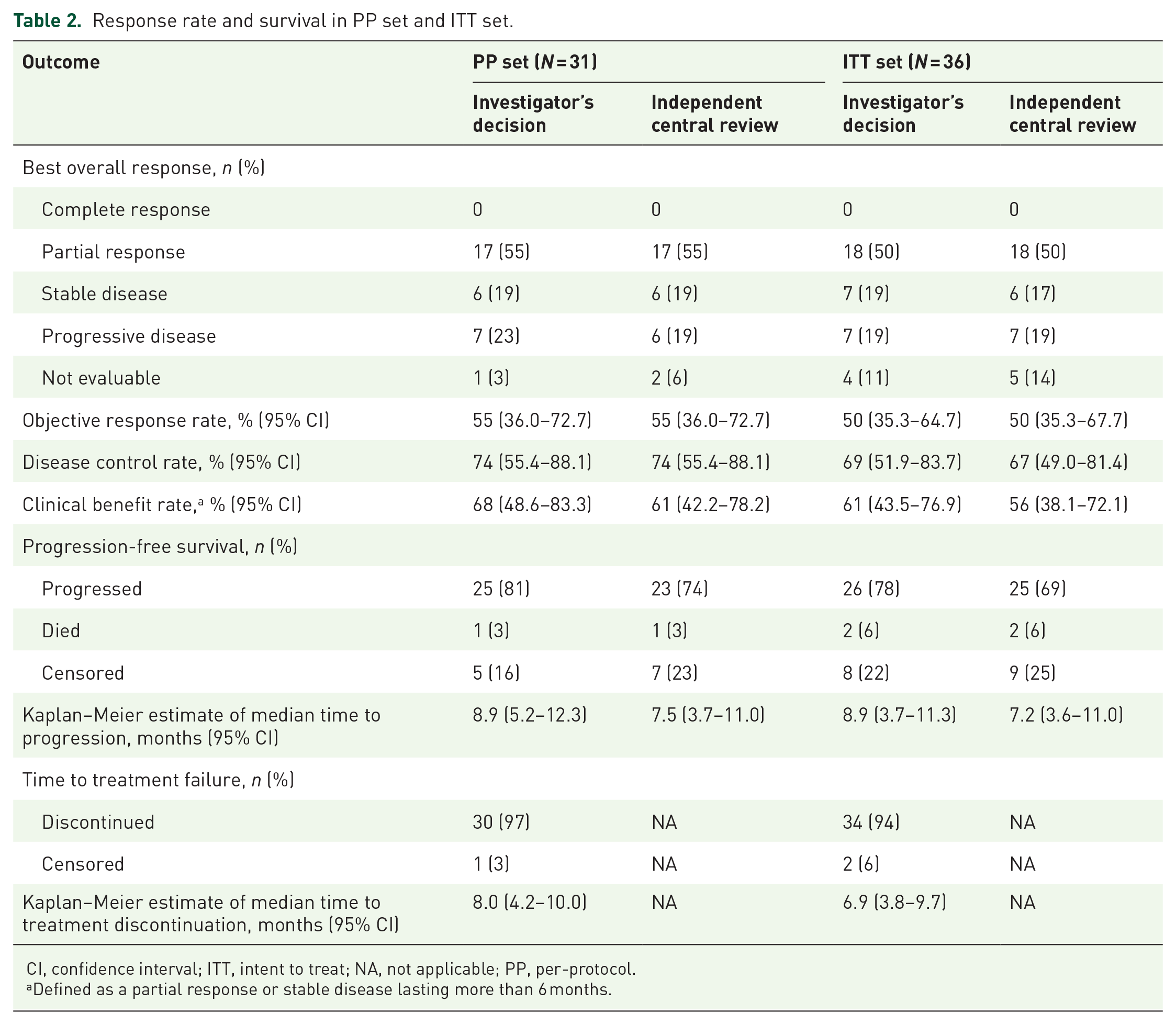
CI, confidence interval; ITT, intent to treat; NA, not applicable; PP, per-protocol.
Defined as a partial response or stable disease lasting more than 6 months.
QoL
Patients’ QoL was maintained during the study. The median EQ-5D-3L score was 0.87 (95% CI: 0.81–0.88; n = 31) at baseline and at end of treatment (95% CI: 0.77–0.87; n = 28). Similarly, the EQ-VAS score was 80.00 at baseline (95% CI: 71.75–82.64; n = 31) and at end of treatment (95% CI: 67.64–80.43; n = 28).
Safety
All 36 patients experienced TEAEs (Table 3). The most common TEAEs were decreased neutrophil count (81%), alopecia (61%), peripheral sensory neuropathy (61%), nausea (56%), and diarrhea (53%). The most common grade ⩾ 3 TEAEs were decreased neutrophil count (78%), anemia (17%), and peripheral sensory neuropathy (8%). There were no cases of febrile neutropenia or sepsis. TEAEs resulted in the dose of DHP107 being interrupted in 26 patients (72%), reduced in 10 patients (28%), and permanently discontinued in four patients (11%). The reasons for permanent discontinuation were grade ⩾ 3 peripheral sensory neuropathy in three patients and ovarian clear-cell carcinoma in one patient. Five patients experienced serious TEAEs; herpes zoster infection, lung abscess (grade ⩾ 3), rib fracture, ovarian clear-cell carcinoma (grade ⩾ 3), and suicide attempt (grade ⩾ 3) each occurred in one patient (3%). Only the herpes zoster infection was considered to be a serious TEAE related to oral DHP107. Serious TEAEs resulted in hospitalization in four patients (11%) and death in one patient (3%), which was due to suicide and deemed unrelated to the study drug.
TEAEs occurring in ⩾ 20% of patients (N = 36).

TEAE, treatment-emergent adverse event.
Peripheral sensory neuropathy occurred in 15 patients up to and including cycle 6 and seven patients after cycle 6.
DHP107 dose and administration
The median number of cycles administered was seven (range = 1–28), with a median treatment duration of 6.3 months (range = 0.5–25.4). Nineteen patients (53%) had a cycle delay, which was due to TEAEs in 18 patients (95%), a schedule change in two patients (11%), and other reasons in three patients (16%). The median total dose of DHP107 administered was 10,600 mg (range = 1,400–41,200), with a median relative dose intensity of 78.5% (range = 38.3–100%). Twenty-five patients (70%) received the full DHP107 dose of 200 mg/m2, and 11 patients had a dose reduction due to TEAEs, seven patients (19%) had a dose reduction to 150 mg/m2, and four patients (11%) had a dose reduction to 112.5 mg/m2. TEAEs leading to dose reduction were grade ⩾ 3 neutrophil count decrease in seven patients (19%), grade ⩾ 3 rash in one patient (3%), grade 2 peripheral edema, grade 2 fatigue, and grade 2 peripheral sensory neuropathy each in one patient (3%). Twenty-six patients (72%) had a dose interruption due to TEAEs, the most common of which was neutrophil count decrease in 23 patients (64%), which was grade ⩾ 3 in 22 patients (61%). Other reasons included nausea in four patients (11%) and stomatitis in two patients (6%).
Discussion
For HR-positive and HER2-negative breast cancer, CDK 4/6 inhibitors, such as abemaciclib, palbociclib, and ribociclib, have changed the treatment paradigm and become a mainstay of therapy for first-line treatment in combination with either an aromatase inhibitor or fulvestrant and second-line treatment in combination with fulvestrant.3,19 However, chemotherapy is still an important option in the treatment armamentarium for HER2-negative breast cancer. Following the positive results for oral DHP107 demonstrated in the phase III DREAM study in gastric cancer, 16 the efficacy and safety of DHP107 were investigated in this phase II study for the first-line treatment of patients with recurrent or metastatic HER2-negative breast cancer. The study met the primary end point, with an investigator-assessed ORR of 55% in the PPS and 44% in patients with triple-negative disease. Efficacy was also demonstrated by the secondary end points, with an investigator-assessed DCR of 74%, median PFS of 8.9 months, and median TTF of 8.0 months. Median OS was not estimable. There was good agreement between the investigator-assessed and ICR efficacy results: the ORR was 55% by both investigator’s decision and ICR, and the DCR was 74% by both investigator’s decision and ICR.
Although the interpretation of the results of cross-study comparison needs to be made with caution, the ORR for oral DHP107 in this study compares favorably with that reported by studies of first-line IV paclitaxel 18 and IV nab-paclitaxel20,21 monotherapy in the treatment of metastatic breast cancer. In a review of studies conducted in the 1990s, Vogel and Nabholtz 18 reported ORRs of 15–62% with IV paclitaxel. Jackisch et al. 21 reported ORRs of 38–49% for studies of weekly IV nab-paclitaxel. A study of IV nab-paclitaxel in routine clinical practice reported an ORR of 9% (one of 11 patients). 20
The efficacy results for oral DHP107 also compare favorably with the results reported for a novel oral formulation of paclitaxel that uses encequidar, a P-glycoprotein pump inhibitor, to enable absorption of oral paclitaxel,22,23 and the oral taxane tesetaxel.24,25 In a phase III study (KX-ORAX-001) comparing oral paclitaxel plus encequidar for 3 days per week versus 3-weekly IV paclitaxel in patients with metastatic breast cancer, the ORR was 40.4% versus 25.6% in the modified ITT population 22 and PFS was 8.4 versus 7.4 months. 23 A phase II study of tesetaxel in patients with HR+/HER2– locally advanced or metastatic breast cancer reported an ORR of 45% and a median PFS of 5.7 months. 25 In the phase III CONTESSA study, in which tesetaxel plus a reduced dose of capecitabine was compared with capecitabine alone in patients with HR+/HER2– metastatic breast cancer who had previously received a taxane, median PFS was 9.8 months for tesetaxel plus capecitabine versus 6.9 months for capecitabine alone. 25 Despite the positive results from these two studies, the development of tesetaxel was discontinued in March 2021 following the decision that the clinical data package was unlikely to receive US Food and Drug Administration approval.
DHP107 had an acceptable tolerability profile. It should be noted that because DHP107 is an oral agent, differences in absorption between individuals can lead to differences in toxicities. The most common TEAEs were decreased neutrophil count (81%), alopecia (61%), and peripheral sensory neuropathy (61%), and the most common grade ⩾ 3 TEAEs were decreased neutrophil count (78%), anemia (17%), and peripheral sensory neuropathy (8%). Peripheral sensory neuropathy occurred in 15 patients up to and including cycle 6 and seven patients after cycle 6. TEAEs led to permanent treatment discontinuation in four patients (11%). There was one death during the study, but this was unrelated to DHP107 treatment.
As response rates in heterogeneous patient populations including Triple-negative breast cancer and HR+ subtypes can vary, we acknowledge that investigator-assessed ORR might not be the most appropriate primary end point in this study. Direct comparison of response rates from one trial to another is inherently difficult, given that studies often differ with respect to entry criteria and population characteristics. In addition, the rate of non-assessable cases [13.9% (5/36)] was relatively high because three patients withdrew in the beginning and two patients were not evaluable due to use of prohibited medication. However, these dropouts mostly occurred at the beginning of the study. It is also important to consider that, while oral treatment with taxanes can be more convenient for patients and improve cost-effectiveness, the use of oral chemotherapy is challenging because of pharmaceutical and pharmacological factors that can lead to low oral bioavailability. Currently, data on the bioavailability of DHP107 in patients from Western countries are lacking, although we hope that an ongoing study on the bioavailability of DHP107 in Caucasian patients (NCT03326102) will help to address this issue.
In conclusion, DHP107 showed promising efficacy with an acceptable tolerability profile in this phase II study and is currently being investigated in the OPTIMAL phase III study with a non-inferiority design (ClinicalTrials.gov Identifier NCT03315364), which includes 476 patients and is being conducted in Korea, China, and Europe (Hungary, Bulgaria, and Serbia). In addition, a phase II clinical trial of DHP107 (OPERA study, ClinicalTrials.gov Identifier NCT03326102) being conducted in the United States and the Czech Republic is evaluating pharmacokinetics, efficacy, and safety.
Footnotes
Acknowledgements
The authors thank the patients, their families, and all the study team members who participated in the OPTIMAL phase II study. They also thank Lee Miller and Kevin De-Voy from Miller Medical Communications for their medical editing/writing assistance, which was funded by Daehwa Pharmaceutical Co., Ltd.
Author contributions
Sung-Bae Kim: conceptualization, methodology, investigation, resources, writing – original draft, writing – review & editing, supervision, and project administration. Jae Hong Seo, Jin-Hee Ahn, Tae-Yong Kim, Seok Yun Kang, Joohyuk Sohn, Yaewon Yang, Kyong Hwa Park, Yong Wha Moon, Seungtaek Lim, and Myoung Joo Kang: investigation and resources. Koung Eun Yoon and Hyun Ju Cho: formal analysis, writing – review and editing, and funding acquisition. Keun Seok Lee: conceptualization, methodology, investigation, and resources.
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S-BK discloses funding from Novartis, Sanofi-Aventis, and DongKook Pharm Co., Advisory Board membership for Novartis, AstraZeneca, Lilly, Daehwa Pharmaceutical Co. Ltd., ISU Abxis, and Daiichi-Sankyo, and holds stock in Genopeaks and Neogene TC. JS discloses funding from MSD, Roche, Novartis, AstraZeneca, Lilly, Pfizer, GSK, Daiichi-Sankyo, Sanofi, and Boehringer Ingelheim. KEY and HJC are employees of Daehwa Pharmaceutical Co. KSL declares personal fees from Roche, Lilly, Novartis, and Dong-A ST. JHS, J-HA, T-YK, SYK, YY, KHP, YWM, SL, and MJK declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Daehwa Pharmaceutical Co., Ltd.