
Abstract
Major breakthroughs have been achieved in the management of metastatic pancreatic ductal adenocarcinoma (PDAC) with FOLFIRINOX (5-fluorouracil + irinotecan + oxaliplatin) and gemcitabine plus nab-paclitaxel approved as a first-line therapy, although the prognosis is still poor. At progression, patients who maintain a good performance status (PS) can benefit from second-line chemotherapy. To address the concern of achieving tumor control while maintaining a good quality of life, maintenance therapy is a concept that has now emerged. After a FOLFIRINOX induction treatment, maintenance with 5-fluorouracil (5-FU) seems to offer a promising approach. Although not confirmed in large, prospective trials, gemcitabine alone as a maintenance therapy following induction treatment with gemcitabine plus nab-paclitaxel could be an option, while a small subset of patients with a germline mutation of breast cancer gene (BRCA) can benefit from the polyadenosine diphosphate-ribose polymerase (PARP) inhibitor olaparib. The rate of PDAC with molecular alterations that could lead to a specific therapy is up to 25%. The Food and Drug Administration (FDA) recently approved larotrectinib for patients with any tumors harboring a neurotrophic tyrosine receptor kinase (NTRK) gene fusion, and pembrolizumab for patients with a mismatch repair deficiency in a second-line setting, including PDAC. Research focused on targeted therapy and immunotherapy is active and could improve patients’ outcomes in the near future.
Keywords
Introduction
Pancreatic ductal adenocarcinoma (PDAC) was the fourth leading cause of death by cancer in the United States and the seventh worldwide in 2019.1,2 Its 5-year survival rate is 9%, which is the lowest of all solid tumors. As its incidence is increasing, PDAC is expected to become the second most common cause of cancer-related death by 2030. 3 Although surgical resection is the only curative treatment, only 15–20% of patients are candidates for surgery. 4 At diagnosis, most patients have a locally-advanced or a metastatic disease. Medical treatment remains the cornerstone for these patients.
In the 1990s, gemcitabine monotherapy became the first-line standard-of-care for metastatic PDAC, after a randomized trial demonstrated its superiority; both in terms of its clinical benefit and overall survival (OS) compared with 5-fluorouracil (5-FU). 5 Major breakthroughs were achieved in 2011 and 2013,6,7 when two large phase III trials showed a clear benefit resulting from a regimen comprising 5-FU, irinotecan and oxaliplatin (FOLFIRINOX) in combination with gemcitabine and nab-paclitaxel, versus gemcitabine monotherapy. The median OS was significantly improved; however, both treatments were associated with higher rates of adverse effects and triggered limiting toxicities. These chemotherapies are now considered as the two main first-line options for patients with metastatic PDAC and a good performance status (PS).
Considering the interesting survival rates provided by these chemotherapies but also their limiting toxicities, the need for maintenance therapy has emerged. Maintenance therapy has recently been evaluated with chemotherapy or targeted therapies. Furthermore, the potential of actionable molecular targets in PDAC patients has garnered significant interest. 8 The Food and Drug Administration (FDA) recently approved the use of pembrolizumab, an anti-programmed-death receptor 1 (anti-PD-1), for patients with any microsatellite instability (MSI) high tumors, including PDAC. Immunotherapy is being studied in larger indications in PDAC.
This review aims to outline the management of metastatic PDAC, from current standard-of-care to recent data on maintenance therapy, and discuss potential future therapeutic directions.
Current standard-of-care
First-line therapy
Until 2011, gemcitabine monotherapy was the standard-of-care in metastatic PDAC. This was based on a randomized trial which showed significant improvement in OS and clinical benefit compared with 5-FU monotherapy. 5 The median OS provided by gemcitabine as a single-agent did not exceed 6 months (5.65 months versus 4.41 months with 5-FU, p = 0.0025) (Table 1).
Clinical trials (randomized phase II and phase III trials) in metastatic PDAC.
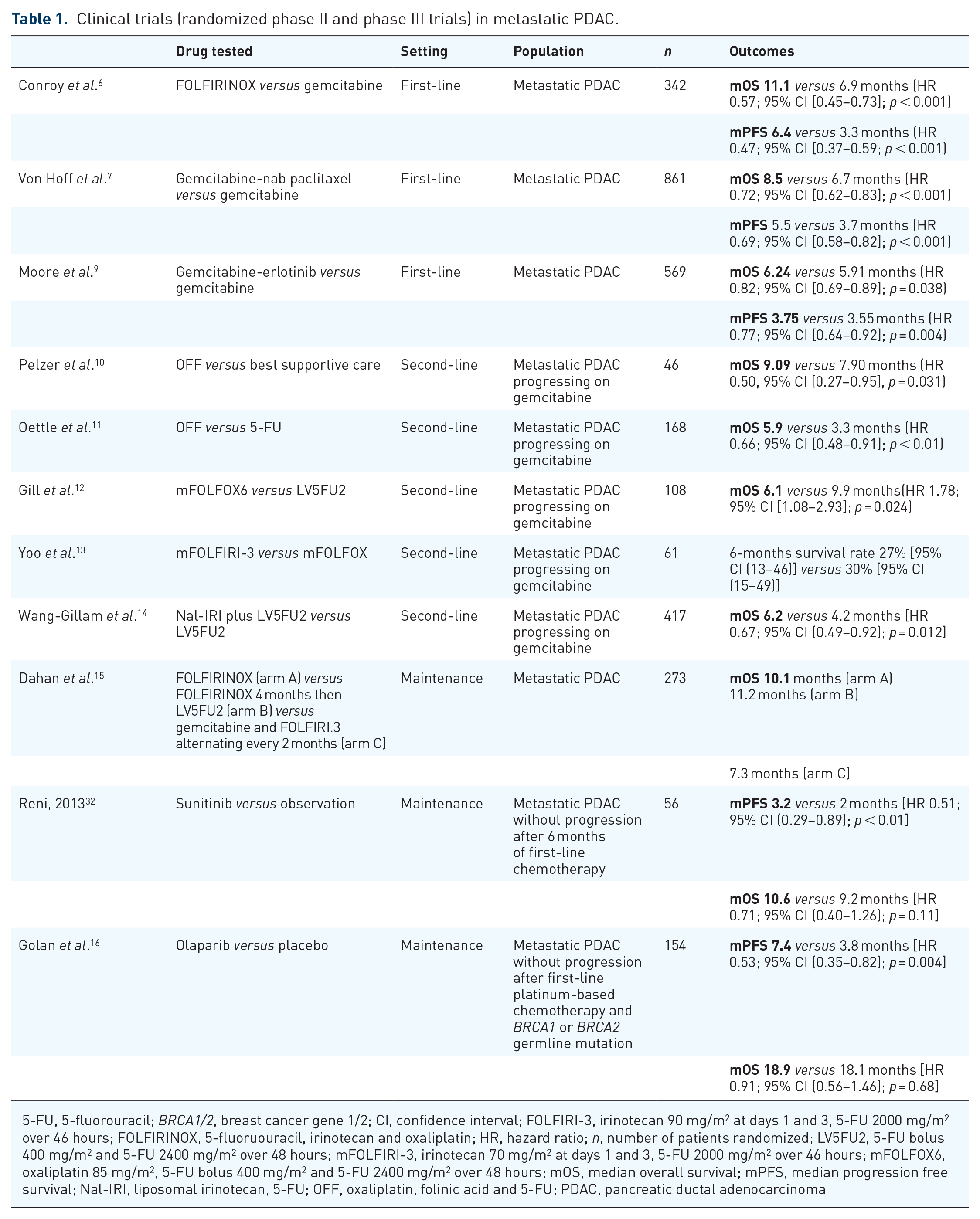
5-FU, 5-fluorouracil; BRCA1/2, breast cancer gene 1/2; CI, confidence interval; FOLFIRI-3, irinotecan 90 mg/m2 at days 1 and 3, 5-FU 2000 mg/m2 over 46 hours; FOLFIRINOX, 5-fluoruouracil, irinotecan and oxaliplatin; HR, hazard ratio; n, number of patients randomized; LV5FU2, 5-FU bolus 400 mg/m2 and 5-FU 2400 mg/m2 over 48 hours; mFOLFIRI-3, irinotecan 70 mg/m2 at days 1 and 3, 5-FU 2000 mg/m2 over 46 hours; mFOLFOX6, oxaliplatin 85 mg/m2, 5-FU bolus 400 mg/m2 and 5-FU 2400 mg/m2 over 48 hours; mOS, median overall survival; mPFS, median progression free survival; Nal-IRI, liposomal irinotecan, 5-FU; OFF, oxaliplatin, folinic acid and 5-FU; PDAC, pancreatic ductal adenocarcinoma
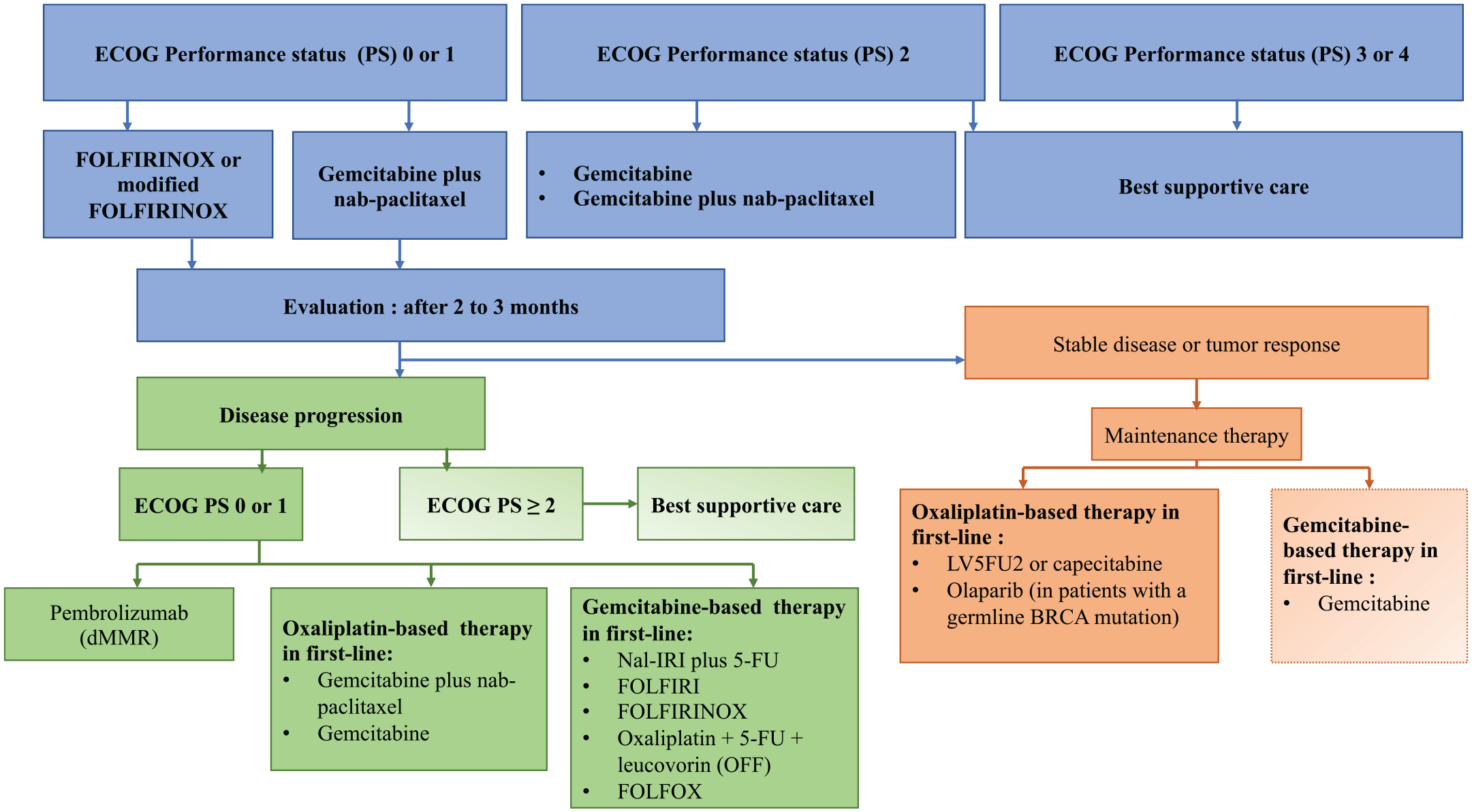
In 2011, a phase III randomized trial compared with the triplet FOLFIRINOX approach to gemcitabine monotherapy and revealed a significant survival benefit of FOLFIRINOX among 342 treatment-naïve patients with an Eastern Cooperative Oncology Group (ECOG) PS of 0 or 1 [median OS 11.1 versus 6.8 months with gemcitabine, 95% CI (0.45–0.73); p < 0.001]. 6 The overall response rate (ORR) and progression-free survival (PFS) were also significantly improved [ORR 31.6% versus 9.4%, PFS 6.4 versus 3.3 months, 95% CI (5.5–7.2)] (Table 1). Since then, FOLFIRINOX became the standard of care (Figure 1). The incidence of grade 3 or 4 neutropenia (45.7%), diarrhea (12.7%), and sensory neuropathy (9%) was significantly higher in the FOLFIRINOX group compared with the gemcitabine group.
Management algorithm proposition for metastatic pancreatic adenocarcinoma.
Considering FOLFIRINOX toxicities, a prospective, single-arm phase II study assessed the efficacy and safety of a modified FOLFIRINOX regimen [oxaliplatin 85 mg/m2, irinotecan 135 mg/m2, 5-FU 300 mg/m2 intravenous (IV) bolus, followed by 2400 mg/m2 continuous infusion for 46 h] in 31 patients with locally advanced and metastatic PDAC. 17 All patients received growth factors. Among patients with metastatic disease, ORR, OS, and PFS appeared similar to results reported by Conroy et al. 6 [ORR 35.1%, OS 10.2 months (95% CI 7.65–14.32) and PFS 6.1 months (95% CI 5.19–8.31)]. Grade 3 or 4 neutropenia and diarrhea occurred in 12.2% and 16.2% of cases, respectively. Another modified FOLFIRINOX regimen (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, 5-FU 2400 mg/m2 infusion, without any bolus of 5-FU) was evaluated in a retrospective study in 60 patients with non-metastatic and metastatic PDAC. The median OS and PFS for metastatic PDAC were 9 months (95% CI 7.1 to not estimable) and 8.5 months (95% CI 3.7–11), respectively. 18 In a large meta-analysis including 32 studies and 1461 patients, there was no difference in OS, PFS, and ORR between FOLFIRINOX and several modified FOLFIRINOX regimens. 19 Although modified FOLFIRINOX regimens have not been compared with standard FOLFIRINOX in a prospective randomized trial, it appears to be a less toxic regimen and numerous data from retrospective studies have shown similar outcomes.
In 2013, a large phase III study randomized 861 patients without any treatment for a metastatic disease and compared with gemcitabine and gemcitabine-plus-nab-paclitaxel. 7 Gemcitabine-plus-nab-paclitaxel was associated with a significant benefit in OS (8.5 versus 6.7 months, HR 0.72, p < 0.001), PFS (5.5 versus 3.7 months, HR 0.69, p < 0.001) and ORR (23% versus 7%, HR 3.19, p < 0.001). The incidence of grade 3 or higher neutropenia, leukopenia, and peripheral neuropathy was significantly higher in the gemcitabine-plus-nab-paclitaxel group (respectively 38%, 31%, and 17%). In contrast to the phase III trial of FOLFIRINOX versus gemcitabine, there was no upper age restriction in this study. Ten percent of patients were at least 75 years of age, and 8% of patients had a Karnofsky PS score between 60% and 70%, corresponding to a PS ECOG of 2.
No prospective randomized trial compared with FOLFIRINOX versus gemcitabine-plus-nab-paclitaxel in a first-line approach. However, retrospective studies did not report any major outcome differences between the two treatment regimens.20–22 In a review of 16 retrospective studies including 3813 patients, the pooled risk of death (HR = 0.99, 95% CI 0.84–1.16, p = 0.9) and progression (HR 0.88, 95% CI 0.71–1.1, p = 0.26) was similar between FOLFIRINOX and gemcitabine-nab paclitaxel; however, the median OS was slightly higher in the FOLRINOX arm [mean weighted OS difference 1.15 months, 95% CI (0.08–2.22), p = 0.03]. 23 Neutropenia and febrile neutropenia occurred significantly more often in the FOLFIRINOX arm. Therefore, gemcitabine-plus-nab-paclitaxel and FOLFIRINOX are both endorsed as first-line therapies by the European Society for Medical Oncology (ESMO) and the American National Comprehensive Cancer Network (NCCN) in patients with metastatic PDAC. A phase I/II trial recently evaluated the combination of liposomal irinotecan, 5-FU, and oxaliplatin (NALIRIFOX) in 32 untreated patients, and reported median PFS and OS of 9.2 (95% CI 7.69–11.69) and 12.6 (95% CI 8.74–18.69) months, respectively. 24 A phase III randomized trial comparing gemcitabine plus nab-paclitaxel to NALIRIFOX in a first-line setting is currently underway (ClinicalTrials.gov identifier: NCT 04083235).
Sequential treatment of gemcitabine-plus-nab-paclitaxel and FOLFIRINOX (GABRINOX) in a first-line setting has been evaluated in a prospective, single-arm phase II trial in 58 patients. 25 The results were promising, with an impressive ORR (63.2%, 95% CI 49.3–75.5) and a prolonged mean OS (mOS) (17.8 months, 95% CI 11.7–21.3). Interestingly, the grade 3 peripheral neuropathy rate was low (5.2%).
The combination of cisplatin and gemcitabine (cisplatin 25 mg/m2, gemcitabine 600 mg/m2) with or without the polyadenosine diphosphate-ribose polymerase (PARP) inhibitor veliparib was evaluated in 50 patients with previously untreated advanced PDAC with a germinal mutation of BRCA/partner and localizer of BRCA (PALB2) in a phase II randomized trial. It revealed an impressive ORR (74.1% in the cisplatin/gemcitabine arm, 65.2% in the cisplatin/gemcitabine-plus-veliparib arm, p = 0.55). 26 The median PFS and OS were also longer than survival rates reported in PDAC trials [median PFS 10.1 months (95% CI, 6.7–11.5), median OS 15.5 months (95% CI, 12.2–24.3) in the cisplatin/gemcitabine arm]. No difference was found in terms of median OS, PFS, and ORR between the two arms. Toxicities in the cisplatin/gemcitabine arm appeared manageable. Therefore, the remarkable ORR achieved with the gemcitabine-plus-cisplatin regimen, combined with its relatively low toxicity, due to doses in this regimen lower than the doses of other indications, makes it a valuable option in the subset of patients with a BRCA/PALB2 germline mutation.
Erlotinib, an inhibitor of the epidermal growth factor receptor (EGFR) tyrosine kinase, is the only targeted therapy to date that has shown, in combination with gemcitabine, a statistically significant improvement in OS in a phase III trial in 2007. 9 A total of five hundred and sixty-nine patients with locally advanced or metastatic PDAC and no prior treatment for metastatic disease were randomized to receive gemcitabine-plus-placebo or gemcitabine-plus-erlotinib. Both the median OS and PFS were significantly higher in the gemcitabine-plus-erlotinib group, although the improvement was modest (median OS 6.24 versus 5.91 months, p = 0.038; PFS 3.75 versus 3.55 months, p = 0.004). Despite FDA approval and NCCN endorsement, the very modest survival improvement achieved by the combination of erlotinib and gemcitabine undermines its potential to be it the most valuable option for first-line treatment of metastatic PDAC. This combination is also not recommended by the 2015 ESMO guidelines.
In metastatic PDAC, chemotherapy has proven its efficacy in terms of median PFS, OS, and clinical benefit. Despite a large number of studies that have evaluated targeted therapies or chemotherapies, the only two regimens which showed a clinically relevant benefit in terms of survival and which are validated worldwide are the triplet with 5-FU, oxaliplatin, and irinotecan (FOLFIRINOX) and the doublet with gemcitabine-plus-nab-paclitaxel.
Maintenance therapy
FOLFIRINOX and gemcitabine-plus-nab-paclitaxel are associated with toxicities that often lead to dose-reductions and limit their long-term use. In a study by Conroy et al., 6 the median number of cycles of FOLFIRINOX administrated was 10. 6 A retrospective study on 2565 patients with metastatic PDAC in 9 European countries found that dose modifications during the treatment course occurred in 21% of cases for FOLFIRINOX and 20.5% of cases for gemcitabine-plus-nab-paclitaxel. 27 These limiting toxicities and the objective of maintaining a good quality-of-life stress the need for maintenance strategies.
Alternating induction with FOLFIRINOX and maintenance with 5-FU has been assessed in the PANOPTIMOX/PRODIGE 35 randomized phase II trial. 15 There were two hundred and seventy-three patients which were randomized in three arms: FOLFIRINOX for 12 cycles (arm A), FOLFIRINOX for eight cycles then 5-FU until disease progression (arm B), or a sequential treatment, alternating between FOLFIRI and gemcitabine every 2 months (arm C). Six month PFS appeared nearly similar in arm A and arm B (47% and 44% respectively) whereas it was lower in arm C (34%). The median OS was also very similar in arm A and arm B (10.1 months 11.2 months respectively), but the 18-month OS rate was higher in arm B (28% versus 18.5%, p < 0.05 in an interim analysis). Surprisingly, the grade 3 or 4 neurotoxicity rate was the highest in the maintenance therapy group (19% versus 10% in arm A), probably because of a higher cumulative oxaliplatin dose after reintroduction in this arm. Capecitabine, an oral prodrug of 5-FU, was evaluated in 2016 in a retrospective study as maintenance therapy after an induction treatment by FOLFIRINOX. 28 Thirty patients without progression after 4-8 cycles of FOLFIRINOX were included. The median PFS on maintenance therapy was 5 months. At reintroduction of the intravenous chemotherapy, the median PFS was 10 months and the median OS was 17 months.
Recently, a retrospective multicentre study evaluated real-life clinical practice of FOLFIRINOX de-escalation and maintenance strategies in 321 patients with metastatic or locally-advanced PDAC. 29 A total of forty-six percent of patients received a maintenance therapy after a median number of nine cycles of FOLFIRINOX. The maintenance therapies were FOLFIRI (45%), oral or 5-FU (35%), and FOLFOX (17%). The median OS for the entire cohort was 16.1 months (95% CI 13.7–20.3). The median OS was similar between those who received 5-FU and those who received FOLFIRI (16.6 versus 18.7 months, p = 0.86), as was PFS (10.1 versus 9.0 months, p = 0.33). Grade 3 or 4 toxicities were more frequent with FOLFIRI maintenance therapy than with 5-FU (41% versus 22%, p = 0.03).
Maintenance with gemcitabine could be interesting after gemcitabine plus nab-paclitaxel induction and has been studied in a prospective study that evaluated maintenance with gemcitabine alone after three cycles of gemcitabine plus nab-paclitaxel in 36 patients aged 70 and over. 30 This study showed a six-month diseases control rate (DCR) of 61% (95% CI, 45–77) and a median OS of 13.4 months (95% CI, 11.1–16.7). The median number of cycles of gemcitabine alone was three (range, 2–9 cycles) and nab-paclitaxel was reintroduced for eight patients after disease progression. Seventeen percent of patients developed sensory neuropathy, although they never reached grade 3. A ‘stop-and-go’ approach, consisting of suspending nab-paclitaxel when patients had grade 3 sensory neuropathy and then reintroducing it at disease progression, was evaluated in a small retrospective study. 31 Here, the continuation of nab-paclitaxel after its reintroduction resulted in a mean second PFS of 2.2 months (range 1–4 months).
In a randomized phase II PACT-12 trial which included 56 patients with stable disease after first-line chemotherapy, sunitinib, an oral multi-targeted tyrosine kinase inhibitor (TKI) targeting the vascular endothelial growth factor receptor (VEGF) and the platelet-derived growth factor receptor (PDGFR), was associated with a significantly longer median PFS than observation [3.2 versus 2 months, HR 0.51; 95% CI (0.29–0.89); p < 0.01]. 32
Olaparib is a PARP inhibitor that has shown efficacy in patients with a germline BRCA mutation and ovarian or breast cancer.33,34 Its efficacy as a maintenance therapy in patients with metastatic PDAC who had not progressed after a platinum-based chemotherapy and a germline BRCA 1 or 2 mutation was recently evaluated in the phase III POLO trial 16 (Figure 2). Of the 3,315 screened patients, only 247 had a germline BRCA mutation, which is consistent with the 4–7% frequency reported in the literature.35–37 A total of one hundred and fifty-seven patients were randomized to receive either olaparib or placebo. The median PFS was significantly improved in the olaparib group [7.4 versus 3.8 months, HR 0.53; 95% CI (0.35–0.82); p = 0.004]. Grade 3 or 4 toxicities occurred in 49% of patients in the olaparib group. Final OS results were recently presented and showed no difference between the two arms (19.0 in the olaparib arm versus 19.2 months in the placebo arm, p = 0.3487), 38 whereas the 36-month OS rates were higher in the olaparib group (33.9% versus 17.8%). The authors highlight that the study was underpowered to detect a difference in OS; even though cross-over was not allowed, 26% of patients in the placebo arm received a PARP inhibitor, which may have biased the results. It should be noted that olaparib was compared with a placebo, although discontinuation of chemotherapy at stability is not the current standard-of-care. Given the use of a placebo as a comparator and the conflicting PFS and OS results, it remains unclear whether olaparib is the most beneficial option for maintenance therapy in patients with a BRCA germline mutation. This also raises the issue of germline testing for patients at diagnosis of metastatic PDAC.

Magnetic resonance imaging of a patient with a metastatic pancreatic adenocarcinoma at baseline (a) and after 6 months FOLFIRINOX chemotherapy (b). After a good response under FOLFIRINOX, the patient underwent maintenance treatment with olaparib.
Maintenance therapy is a daily necessity and appears feasible for example with
LV5FU2 (5-FU bolus 400 mg/m2 and 5-FU 2400 mg/m2 over 48 hours) after at least 8–12 cycles of FOLFIRINOX. Gemcitabine alone after induction treatment with gemcitabine-plus-nab-paclitaxel could also reduce limiting toxicities but probably requires further validations by larger studies. The optimal number of cycles of combination chemotherapies remains an issue.
Second-line therapy
In the FOLFIRINOX versus gemcitabine trial, only 46.7% of heavily-selected patients underwent a second-line treatment at progression. 6 In a meta-analysis that included 71 second-line studies and 3,112 patients, active treatment compared with the best supportive care (BSC) showed a significantly longer median OS (4.6 versus 2.5 months respectively, p = 0.02). 39 Retrospectives studies tried to identify prognostic factors to select patients that could benefit from a second-line therapy. In a retrospective analysis of 90 patients, an ECOG PS of 0 or 1, an albumin level of at least 35 g/l, and response from a first-line therapy seemed associated with a better outcome regarding OS. 40 In another retrospective analysis of 280 patients who received second-line treatment for PDAC, 41 Karnofsky PS, carbohydrate antigen 19-9 (CA 19-9) at the beginning of second-line therapy, and the duration of first-line treatment significantly influenced median OS.
In 2009, the alkylating agent glufosfamide failed to improve OS compared with BSC in a second-line setting in a phase III trial. 42
In 2011, a phase III trial compared with BSC alone versus an
OFF regimen (folinic acid 200 mg/m2, 5-FU 2000 mg/m2 over 24 jours on days 1, 8, 15, 22 and oxaliplatin 85 mg/m2 on days 8 and 22) for patients with advanced PDAC who experienced progression during gemcitabine therapy. 10 The OFF regimen consisted of a 6 weeks cycle of folinic acid and 5-FU administrated on days 1, 8, 15, and 22, and oxaliplatin administrated on days 8 and 22. The trial was closed due to insufficient accrual after the inclusion of 46 patients; however, the median second-line survival and median OS were significantly longer in the OFF group [4.82 versus 2.30 months, p = 0.008, HR 0.45, 95% CI (0.24–0.83) and 9.09 versus 7.90, p = 0.031, HR 0.50, 95% CI (0.27–0.95) respectively]. The OFF regimen was later compared with folinic acid and fluorouracil in the CONKO-003 phase III trial which included 168 patients. 11 The median OS was improved in the OFF group [5.9 versus 3.3 months, HR 0.66, 95% CI (4.1–7.4)] as well as the median time to progression [2.9 versus 2.0 months, HR 0.68, 95% CI (0.50–0.94), p = 0.019]. Data from 27 patients who received FOLFOX as second-line treatment among patients included in the FIRGEM trial, which compared with gemcitabine alone to gemcitabine alternating with FOLFIRI3 as a first-line treatment, were prospectively analyzed in an observational cohort study. 43 The median OS and PFS from the start of the second-line therapy were 4.3 and 1.7 months, respectively. In contrast, the addition of oxaliplatin to 5-FU reduced the OS in the PANCREOX phase III trial. 12 A total of one hundred and eight patients were randomized to receive either LV5FU2 (bolus of 5-FU followed by a 2400 mg/m2 continuous infusion) or biweekly
mFOLFOX6 (bolus of 5-FU 400 mg/m2 followed by a continuous infusion of 2400 mg/m2 and oxaliplatin 85 mg/m2). While the ORR and PFS did not significantly differ between the two groups, the median OS was significantly lower in the mFOLFOX6 group [6.1 versus 9.9 months, HR 1.78; 95% CI (1.08–2.93), p = 0.024]. This lack of benefit may be explained by greater toxicity in the mFOLFOX6 arm, with more patients withdrawn from the study due to adverse events (63% versus 11%), and the more frequent use of subsequent treatment lines after progression in the LV5FU2 arm (23% versus 7%). Therefore, the data on the combination of 5-FU and platinum as second-line approaches are controversial.
FOLFIRI-1 (irinotecan 180 mg/m2, leucovorin 400 mg/m2, 5-FU 400 mg/m2 bolus, then an infusion of 2400 mg/m2) and FOLFIRI-3 (irinotecan 100 mg/m2 on days 1 and 3, leucovorin 400 mg/m2, and an infusion of 2400 mg/m2 of 5-FU without bolus) regimens were evaluated in a prospective cohort of 63 patients as a second-line therapy for metastatic PDAC resistant to gemcitabine and platinum-salts. 44 The DCR was 39.7% while the median OS was 6.6 months. The FOLFIRI regimen has also been evaluated in a phase II trial comparing a modified FOLFIRI-3 regimen (irinotecan 70 mg/m2 on days 1 and 3, leucovorin 400 mg/m2, and 5-FU 2000 mg/m2) to mFOLFOX in 61 patients with gemcitabine-refractory advanced PDAC. 13 The DCR [23% in the mFOLFIRI group, 95% CI (11–40) and 17% in the mFOLFOX group, 95% CI (7–34)] and 6-month survival rates [27% in the mFOLFIRI group, 95% CI (13–46) and 30% in the mFOLFOX group, 95% CI (15–49)] were almost identical, as well as grade 3 or 4 toxicities.
In a retrospective analysis of 181 patients who progressed on gemcitabine-plus-nab-paclitaxel, mOS and mPFS were higher in patients treated with a platinum-based doublet than those treated with an irinotecan-based doublet (free irinotecan or liposomal irinotecan). However, this did not reach statistical significance (mOS 10.3 versus 8.2 months, p = 0.713; mPFS 4.0 versus 3.3 months, p = 0.494). 45
A liposomal formulation of irinotecan (nal-IRI) was tested as a second-line therapy after a gemcitabine-based first-line in the phase III NAPOLI-1 trial.14,46 A total of four hundred and seventeen patients were randomized into three groups: nal-IRI monotherapy, nal-IRI plus LV5FU2, or LV5FU2 monotherapy. The median OS was significantly longer in the nal-IRI plus LV5FU2 group, as compared with LV5FU2 monotherapy [6.2 versus 4.2 months, HR 0.67, 95% CI (0.49–0.92); p = 0.012]. There was no difference in median OS between the nal-IRI monotherapy group and the LV5FU2 group. The ORR was higher in the nal-IRI plus LV5FU2 group than in the LV5FU2 monotherapy group (17% versus 1%, p = 0.0001). Nal-IRI plus LV5FU2 is currently recommended by ESMO (European Society for Medical Oncology) and NCCN guidelines for patients who have progressed after a gemcitabine-based first-line therapy. It should be noted that Nal-IRI is not available in many parts of the world, and, while it has been shown in preclinical studies to achieve higher intratumoral levels of irinotecan and its active metabolite SN-38, 47 no randomized trial has compared Nal-IRI-plus-5-FU with FOLFIRI. Interestingly, no differences in OS and PFS were found in a retrospective study which compared Nal-IRI/5FU with FOLFIRI in patients with advanced PDAC who had received prior gemcitabine-based chemotherapy (mOS 7.1 versus 6.7 months, mPFS 4.1 versus 3.1 months). 48 Similar rates of grade 3 or 4 toxicities were observed between the two groups.
No prospective randomized studies on FOLFIRINOX or modified FOLFIRINOX as a second-line therapy have been conducted. One retrospective study of 27 patients that had progressed after a gemcitabine monotherapy found a median OS of 8.5 months and a DCR of 63%. 49 Similar findings were reported in another retrospective study conducted on 18 patients. 50 Recently, retrospective analysis on 52 patients who had progressed after gemcitabine-plus-nab-paclitaxel and underwent a second-line with a modified FOLFIRINOX identified a median OS of 22.5 months for metastatic disease. 51 A modified FOLFIRINOX (oxaliplatin 65 mg/m2, irinotecan 135 mg/m2, 5-FU 2000 mg/m2 continuous infusion with no bolus) has been evaluated in a prospective single-arm Korean trial in 39 patients who had progressed after gemcitabine-based chemotherapy. 52 The median OS and PFS were 8.5 months (95% CI 5.6–11.4) and 3.8 months (95% CI 1.5–6), respectively. In daily practice, triplet chemotherapy in a second-line setting is often perceived as too toxic.
Several retrospective studies have evaluated gemcitabine as a single agent in second-line therapy after FOLFIRINOX failure,53–56 and reported a median OS from 3.6 to 5.7 months. Recently, a phase II trial assessed the efficacy and safety of the combination of gemcitabine-plus-nab-paclitaxel after FOLFIRINOX in 30 patients under 75 years-old with an ECOG PS of 0 or 1. 57 The ORR and DCR were 13.3% and 46.7%, respectively. The median OS from the beginning of the second-line treatment was 7.6 months. Similar results were found in a prospective cohort of 57 patients who received gemcitabine/nab-paclitaxel after FOLFIRINOX failure (ORR 17.5%, DCR 58%, median OS 8.8 months). 58 In a retrospective study in 103 patients with advanced PDAC who received gemcitabine-plus-nab-paclitaxel after FOLFIRINOX failure, the mOS and mPFS were 9.8 months and 4.6 months, respectively. 59 In a recent meta-analysis including 16 studies, the DCR was higher for patients treated with gemcitabine-plus-nab-paclitaxel compared with single-agent gemcitabine (53.5% versus 30.2%, p < 0.001) after FOLFIRINOX failure. 60 Although the vast majority of available data are from retrospective trials, gemcitabine-plus-nab-paclitaxel in selected patients who have progressed after platinum-based therapy seems to be associated with improved outcomes when compared with gemcitabine monotherapy.
The use of a second-line treatment could benefit selected patients who maintain a good general condition. For these patients, nal-IRI plus 5-FU is currently endorsed by American and European guidelines. Although data on FOLFIRINOX in a second-line setting are limited, it could be an option, as well as gemcitabine-plus-nab-paclitaxel after platinum-based first-line therapy. One should note that clinical practices regarding the choice of combination chemotherapy in the first- or in the second-line setting is currently affected by national regulations and drug reimbursement policies.
Future directions
It is estimated that the rate of pancreatic tumors with molecular alterations for which there is clinical or preclinical evidence of a benefit from a specific therapy is up to 25%.61–65
Recently, tumor samples from 1028 patients referred to the Know Your Tumour program, a program of molecular profiling for patients with PDAC launched by the Pancreatic Cancer Action Network (PanCAN), were analyzed. 8 A total of one hundred and eighty-nine patients out of 617 had actionable molecular alteration and 46 patients received a molecularly-matched treatment. The median OS was significantly longer in the matched therapy group than in the group of patients who had molecular alterations and who received unmatched therapy [2.58 versus 1.51 years; HR 0.42; 95% CI (0.26–0.68), p = 0.0004].
KRAS pathway
Mutations in the Kirsten rat sarcoma viral oncogene homology (KRAS) oncogene are found in more than 90% of PDAC. 64 It is well-demonstrated that KRAS mutation predicts resistance to EGFR targeted-therapy in colon cancer,66,67 whereas its implication in PDAC is more controversial.
Erlotinib, an inhibitor of EGFR tyrosine kinase, has shown a statistically significant but modest improvement on OS in combination with gemcitabine for metastatic PDAC, in the NCIC CTG PA.3 phase III trial, but KRAS mutation was not analyzed. 9 In a retrospective study of 136 patients with locally-advanced or metastatic PDAC who received first-line gemcitabine-based chemotherapy with or without erlotinib, the median OS was significantly longer in the subgroup of KRAS wild-type patients treated with gemcitabine-erlotinib (9.7 versus 5.2 months, p = 0.002). 68 There was no difference in OS based on the KRAS mutation status in the subgroup of patients treated with gemcitabine without erlotinib (7.0 versus 7.0 months; p = 0.121). In contrast, the retrospective analysis of 146 tumor samples from patients included in the NCIC CTG PA.3 trial did not reveal any significant interaction between KRAS mutation status and OS in patients treated with gemcitabine/erlotinib. 69 Similarly, no association between KRAS mutation status and outcomes was found in a prospective randomized Taiwanese trial evaluating gemcitabine with or without erlotinib in 88 treatment-naive patients. 70 In contrast, in patients treated with gemcitabine-plus-erlotinib, the mOS, mPFS, and DCR were significantly higher in those with EGFR mutations than in those with no EGFR mutations (mOS 8.7 versus 6.0 months, p = 0.044; mPFS median 5.9 versus 2.4 months, p = 0.004; DCR 85% versus 33%; p = 0.001). Therefore, data on KRAS and EGFR mutations status as predictive biomarkers for inhibitor of EGFR tyrosine kinase efficacy are uncertain and require more evaluation.
Cetuximab, an anti-EGFR monoclonal antibody, failed to improve survival in combination with gemcitabine in a phase III, which was not stratified according to KRAS mutation status. 71
Nimotuzumab, another anti-EGFR monoclonal antibody, has shown efficacy in combination with gemcitabine in a first-line setting for OS and PFS in a phase IIb randomized, placebo-controlled trial including 192 patients. 72 Both median OS and PFS were significantly higher in the gemcitabine-plus-nimotuzumab group (median OS 8.6 versus 6.0 months; HR 0.69; p = 0.03; median PFS 5.1 versus 3.4 months; HR 0.68; p = 0.02). At 12 months, patients with a KRAS wild-type status had a significantly better OS rate in the nimotuzumab group than in the placebo group (53.8% versus 15.8%; HR 0.32; p = 0.026).
Other approaches to target KRAS by inhibiting its downstream signaling pathways, such as the Ras-Raf-MEK-ERK pathway, have been attempted. The addition of trametinib, a selective mitogen-activated protein kinase (MEK) inhibitor, to gemcitabine did not improve OS in a phase II trial. 73 Selumetinib, another MEK inhibitor, also failed to improve OS in comparison with capecitabine in a second-line setting. 74
KRAS encodes for a guanosine triphosphatase (GTPase) protein, which cycles between an inactive guanoside diphosphate (GDP)-bound state and an active guanosine triphosphate (GTP)-bound state. Oncogenic mutations of KRAS impair GTPase protein activity, resulting in the accumulation of its active form. The KRAS G12C mutation is rare in PDAC (1% of PDAC), 75 but recently, small molecules that specifically and irreversibly inhibit the KRASG12C mutation have garnered interest. KRASG12C inhibitors lead to the trapping of the mutant KRAS protein in its inactive GDP-bound state, and have shown durable tumor regression in preclinical studies.76,77 The KRASG12C inhibitor Sotorasib (AMG 510) showed promising activity in a phase I clinical trial in 129 pre-treated patients including 12 PDAC. 78 Among the 28 patients with other tumor types, 4 had a partial confirmed response (including one with PDAC) while 17 had stable disease. A large phase Ib trial evaluating sotorasib as a single agent or in combination for patients with advanced solid tumors with KRASG12C mutation is currently ongoing (ClinicalTrials.gov identifier: NCT04185883).
PARP inhibitors
The BRCA 1 and 2, ATM serine/threonine kinase (ATM), PALB2, ATR serine/threonine kinase (ATR), and fanconi anaemia complementation group A (FANC) genes are involved in homologous recombination (HR) repair mechanisms which play a key role in repairing double-strand breaks (DSB) of DNA. Approximately four-to-seven percent of patients with PDAC harbor a germline mutation of BRCA 1 or 2.35–37 PARP is involved in repairing single-strand DNA breaks (SSB) and its inhibition results in the accumulation of SSB and eventually conversion to DSB, which in case of HR deficiency leads to cell death.
Olaparib monotherapy was evaluated in a phase II trial involving 298 patients who progressed on prior chemotherapy with advanced cancer and a germline BRCA mutation, including 23 PDAC. 79 The ORR was 21.7% in PDAC (95% CI, 7.5–43.7) and the median PFS was 4.6 months. No difference was found concerning ORR between patients who did or did not receive prior platinum-therapy.
The efficacy of rucaparib, another PARP inhibitor (PARPi), as a single agent in patients with metastatic PDAC who received one or two previous lines of chemotherapy, has been investigated in a phase II trial conducted on 19 patients with germline (n = 16) or somatic (n = 3) BRCA mutations. 80 Enrolment was stopped because the study did not meet its primary endpoint, with a DCR of 32% and an ORR of 15.8%. Interestingly, none of the four tumors of the patients who responded to rucaparib had progressed with prior platinum therapy, which is consistent with previous findings regarding platinum sensitivity conferred by DNA-damage repair (DDR) mutations. 65 A retrospective analysis of patients with advanced PDAC who received platinum-based therapy in a first-, second-, or third-line setting found a significantly higher ORR in patients harboring a BRCA or PALB2 mutation, in comparison with control patients (58% versus 21%, p = 0.0022), as well as a longer median PFS (10.1 months versus 6.9 months, p = 0.0068). 81
Veliparib was tested in monotherapy in a phase II trial including 16 patients with pre-treated PDAC and a germline BRCA mutation. 82 No confirmed radiological response was observed. It should be noted that 88% percent of patients had previously been treated with platinum-based therapy, while 64.3% of them experienced disease progression while on this regimen. Recently, a phase I/II study of veliparib, in combination with FOLFOX for previously untreated or treated metastatic PDAC patients with HR genes mutation or a suggestive family history, showed an ORR of 26%. 83 In the subgroup of patients without prior platinum-based therapy, ORR and DCR were 32% and 64% respectively, while the ORR for previously platinum-treated patients was 7% and DCR was 14%. The highest ORR in this study was found in the subgroup of patients with an HR gene mutation who had not been previously exposed to platinum salts (ORR 57%). This raises the issue of the association between platinum-sensitivity and response to PARPi’s in patients displaying disease progression under prior chemotherapy, as restoration of the HR system has been described and could be a resistance mechanism to platinum and PARPi therapy. 84 The addition of veliparib to the combination of gemcitabine and cisplatin in a first-line setting did not improve ORR, median PFS, and OS in a phase II randomized trial for previously untreated patients with an advanced PDAC and a BRCA/PALB2 germline mutation. 26 Therefore, the contribution of veliparib to platinum-based therapy in a first-line setting for patients harboring a BRCA or PALB2 mutation is uncertain.
Olaparib has proven its efficacy as a maintenance therapy for metastatic PDAC that had not progressed during first-line platinum-based chemotherapy in patients with a germline BRCA mutation. 16 However, the place of PARPi’s as maintenance therapy is not clearly-defined, as it has not been compared with cytotoxic chemotherapy in this setting. Moreover, it is still unclear whether PARPi could benefit patients with somatic mutation in the HR genes. Rucaparib is currently being tested (ClinicalTrials.gov identifier: NCT03140670) (Table 2) as maintenance therapy in a single-arm phase II study in patients with a somatic or germline mutation in BRCA 1, BRCA 2, or PALB2 and a platinum-sensitive PDAC. Early results are encouraging, with a DCR of 89.5%. 85 A phase II study evaluating olaparib as a maintenance therapy in patients with a somatic BRCA mutation has recently started (ClinicalTrials.gov identifier: NCT04348045) (Table 2).
Ongoing clinical trials on immunotherapy and targeted therapy for metastatic pancreatic adenocarcinoma.

Status available on October 2020.
ALK, anaplastic lymphoma kinase; BRCA, breast cancer antigen; CAR-T, carbonic anhydrase 1; CLD18, claudin 18.2; EGFR, epidermal growth factor receptor; KRAS, Kirsten rat sarcoma viral oncogene homolog; MEK, mitogen-activated protein kinase; NRG1, neurogulin 1; PALB2, partner and localizer of BRCA2; PARP, poly ADP ribose polymerase; PDAC, pancreatic ductal adenocarcinoma; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; PSCA, prostate stem cell antigen.
PARPi’s are a promising addition to metastatic PDAC drugs for patients with a BRCA mutation, but many questions remain, such as the appropriate timing of their use and their efficacy in patients with a somatic BRCA mutations.
Other explored pathways
Among the small, but non-negligeable proportion of PDAC with no KRAS mutation (8–12%62,65), several molecular alterations are therapeutically targetable. These include the fusion in the neurotrophin receptor tyrosine kinase (NRTK) and ROS1 fusions, NRG1 fusions, ALK rearrangement and BRAF mutations, have been found to be more prevalent than in KRAS mutated PDAC.86–89
Fusions in the NRTK gene family leads to a chimeric tropomyosin receptor kinase (TRK) fusion protein and is an oncogenic driver in various types of tumors. Larotrectinib, a TRK inhibitor, has shown efficacy in a phase I–II trial including 1 PDAC, with an ORR of 75%, 90 and is approved by the FDA for any tumor harboring an NTRK gene fusion. Entrectinib, another TRK-inhibitor showed an ORR of 57% (95% CI, 43.2–70.8%) in an analysis of 3 phase I–II trials with 3 PDAC. 91 Fusions in the NTRK family gene is rare (<1% in every solid tumor 92 ), but given the benefits from TRK inhibitors, molecular testing for NTRK gene fusions could be considered in patients with advanced PDAC and an ECOG PS of 0 or 1 without any other therapeutic proposition.
Translocation of the anaplastic lymphoma Kinase (ALK) gene results in constitutive activation of ALK, which encodes for a TRK protein and has been described in various solid tumors. 93 Genomic profiling of 3,170 locally advanced or metastatic PDAC identified 5 ALK-rearrangement tumors (0.16%) in young patients (<50 years old). 87 The ongoing basket trial MATCH (ClinicalTrials.gov identifier: NCT02465060) (Table 2) will evaluate crizotinib, an ALK protein inhibitor, among other therapies for various tumors.
Gene fusions involving neuregulin-1 (NRG1) activate the human epidermal growth factor (HER2-HER3) receptor tyrosine kinase pair, leading to the pathological activation of its downstream signaling pathway, such as phosphoinositide 3-kinase (PI3K)/ protein kinase B (AKT). The HER-family kinase inhibitor afatinib has shown some efficacy for these tumors.94,95 In a whole-genome and transcriptome sequencing of 47 PDAC, Jones et al. detected three patients with NRG1 fusion-positive tumors, all of them KRAS wild-type. 96 Two of them were treated with afatinib. Both had a radiologic response at 3 months. One experienced disease progression 5 months after initiation of afatinib. A phase II trial (NCT04410653) (Table 2) of afatinib for advanced solid NRG1-rearranged tumors, including PDAC, is scheduled to start shortly. Zenocutuzumab, a bispecific monoclonal antibody that binds to HER 2 and HER 3 receptors and blocks the interaction of NRG1 fusion protein with its receptor HER3, has been shown to induce a response in tumors harboring NRG1 fusions in preclinical and early clinical studies.97,98 A phase I/II trial evaluating zenocutuzumab in patients with various solid tumors harboring a NRG1 fusion is currently underway (ClinicalTrials.gov identifier: NCT02912949).
The mitochondrial metabolism in tumor cells has been shown to be particularly important for PDAC cells. 99 Devimistat (CPI-613) targets the mitochondrial tricarboxylic acid (TCA) cycle, through the inhibition of two of its major enzyme complexes, pyruvate dehydrogenase complex (PDC) and α-ketoglutarate dehydrogenase (KGDH). Encouraging results of a phase I study on CPI-613 combined with FOLFIRINOX with an ORR of 61% (95% CI, 36–83%) 100 led to the start of the AVENGER phase III trial, which evaluated modified FOLFIRINOX with or without CPI-613 (ClinicalTrials.gov identifier: NCT03504423) in the first-line setting. 101 The combination of CPI-613 and gemcitabine-plus-nab-paclitaxel was also evaluated in a phase I trial, resulting in 50% response rate. 102 Therefore, early clinical trials suggest the efficacy of CPI 613 in combination with chemotherapy, but these findings require further confirmations.
Immunotherapy
High microsatellite instability (MSI-h) or mismatch repair deficiency (dMMR) is a rare occurrence in PDAC (~ 0.5–1%103–105). MSI-high tumors, characterized by the strong expression of tumor neoantigens and immune check-point ligands, have been shown to benefit from immune checkpoint inhibitors. 106 The anti-PD1 pembrolizumab has been approved by the FDA for all MSI-h/dMMR tumors, and is endorsed by the NCCN in a second-line setting. In a next-generation sequencing (NGS) of 833 patients with PDAC, MSI-h/dMMR tumors were found in seven patients (~0.84%), of whom five were treated with anti-PD1 or anti-PDL1. Four of them had a response or a stable disease. 105 The KEYNOTE-158 phase II study assessed the efficacy of pembrolizumab for non-colorectal MSI-h/dMMR tumors in a second-line setting or over. 107 Two hundred and thirty-three patients with 27 tumor types, including 22 patients with PDAC, were included. The median OS and PFS were 23.5 months (95% CI, 13.5 months to not reached) and 4.1 months (95% CI, 2.4 to 4.9 months) respectively, while the ORR was 34.3% (95% CI, 28.3-40.8%). However, the ORR [18.2%, 95% CI (5.2–40.3)], median OS [4 months, 95% CI (2.1–9.8)], and median PFS [2.1 months, 95% CI (1.9–3.4)] were lower for PDAC patients than for the entire cohort.
Anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA4) has also been evaluated in PDAC. In a phase II trial, 27 patients with advanced PDAC received ipilimumab but with no objective response. 108 A randomized phase II trial of the anti-PD-L1 durvalumab with or without the anti-CTLA4 tremelimumab in a second-line setting was also disappointing, with an ORR of 3.1% (95% CI, 0.008–16.22) for combination therapy and 0% (95% CI, 0.00–10.58) for monotherapy. 109
The combination of immune checkpoint inhibitors and cytotoxic chemotherapy for metastatic PDAC has also been investigated in phase I studies. While a phase II study evaluating pembrolizumab in combination with either gemcitabine-plus-nab-paclitaxel or FOLFIRINOX is underway (ClinicalTrial.gov identifier: NCT04447092) (Table 2), the addition of tremelimumab and durvalumab to gemcitabine and nab-paclitaxel did not improve OS, PFS, and ORR in the CCTG PA.7 phase II randomized trial. 110
Several retrospective studies have demonstrated an association between tumor mutational burden (TMB), a quantitative assessment of the number of somatic mutations within a tumor genome, and response to immune checkpoint blockade, notably in melanoma and non-small cell lung cancer (NSCLC).111–113 Plasma TMB of patients included in the CCTG PA.7 were analyzed retrospectively. In patients with high-TMB (with a threshold of ⩾9 mut/Mb), there was a trend toward a decreasing HR, favoring the combination of chemotherapy and immunotherapy (HR 0.30, 90% CI 0.06–1.37), in contrast to patients in the low TMB subgroup (HR 0.97, 90% CI 0.73–1.29). 114 It should be noted that only 4.6% of patients had a plasma TMB ⩾ 9 mut/Mb. High-TMB may be a predictive biomarker of tumor response to checkpoint inhibition for metastatic PDAC, but these data require prospective confirmation.
Cancer vaccines have also been investigated in PDAC, with mixed results. 115 To date, the largest study evaluating the vaccines GVAX and CRS 207 in PDAC was disappointing, with no difference in OS with or without GVAX. 116 The addition of nivolumab to GVAX/CRS 207 did not improve OS in a phase II randomized trial in pre-treated PDAC patients. 117 A randomized phase II trial will assess the efficacy of GVAX and CRS 207 in combination with dual immune checkpoint inhibition (ClinicalTrials.gov identifier: NCT03190265) (Table 2). Another area of development is chimeric antigen receptor (CAR)-T cells therapy, which uses genetically-engineered T cells directed to specific cancer-associated antigens. Results from preclinical and phase I studies in PDAC are promising,118–120 and several phase I/II trials on CAR-T cells for various solid tumors, including PDAC, are currently ongoing (ClinicalTrial.gov identifiers: NCT02744287, NCT03159819) (Table 2).
Immunotherapy as a maintenance therapy for metastatic PDAC is an option currently being evaluated in different trials (Table 2).
One possible explanation for the difficulty in achieving a significant and sustained response from immunotherapy for PDAC is the abundance of an immunosuppressive stroma through cancer-associated fibroblast activation and a relatively low level of neoantigens compared with other solid tumors. Future perspectives for immunotherapy will probably combine checkpoint inhibitors with other drugs, or remodel the PDAC microenvironment via depletion of pro-tumorigenic immune cells. 121
Conclusions
Undisputable progress has been made in the management of metastatic PDAC, with a significant subset of patients now achieving a stabilization or a response with combination chemotherapies in the first-line management of their disease. As survival increases slowly and because quality of life is an absolute priority, the concept of maintenance therapy has emerged for metastatic PDAC. Continuing low-dose cytotoxic chemotherapy is a valid option, but switching to PARP inhibitors should be considered in patients with a germline mutation of BRCA 1 or 2. The question of whether PARP inhibitors could benefit patients with a somatic mutation of BRCA 1 or 2 remains a concern that future trials may answer. Besides maintenance therapy, research is ongoing, and a better understanding of the unique characteristics inherent to PDAC and its immunosuppressive microenvironment should shortly lead to the development of novel agents that specifically target signaling pathways and genetic alterations present in patients with PDAC and could achieve substantial survival benefits.
Footnotes
Conflict of interest statement
L-J.P. has conflicts of interest with Servier, Amgen, Merck, MSD, Keocyt. S.D. has conflicts of interest with Mylan, Amgen. C.B. has conflicts of interest with IPSEN. S.C. has conflicts of interest with AAA. R.C. has conflicts of interest with AAA, Bayer, Servier, Ipsen, Novartis and Keocyt. All other authors have no conflict of interest to declare.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.