
Abstract
Background:
The C-TASK-FORCE phase I/II and Danish randomized phase II trials reported the promising efficacy of trifluridine/tipiracil (TAS102) plus bevacizumab (BEV) in patients with chemorefractory metastatic colorectal cancer (mCRC). However, there had been no direct comparative phase III trial to compare the efficacy between TAS102 plus BEV and standard therapy with either TAS102 or regorafenib monotherapy.
Methods:
We retrospectively reviewed the medical records of patients with mCRC who received TAS102 plus BEV, TAS102 monotherapy, or regorafenib monotherapy after standard chemotherapies during 2013–2019.
Results:
Patients received TAS102 plus BEV (n = 139), TAS102 monotherapy (n = 153), or regorafenib monotherapy (n = 133). With a median follow-up of 25.3 months, median overall survival (OS) was 11.5 months [95% confidence interval (CI), 9.9–13.9] for TAS102 plus BEV, 8.1 months (95% CI, 6.8–9.2) for TAS102 monotherapy, and 6.8 months (95% CI, 5.7–8.5) for regorafenib monotherapy. The hazard ratios were 0.67 (95% CI, 0.51–0.88) for TAS102 plus BEV versus TAS102 monotherapy and 0.71 (95% CI, 0.54–0.94) for TAS102 plus BEV versus regorafenib monotherapy. Median progression-free survival (PFS) was 4.4 months (95% CI, 3.7–5.4) for TAS102 plus BEV, 2.5 months (95% CI, 1.6–2.3) for TAS102 monotherapy, and 2.1 months (95% CI, 1.6–2.3) for regorafenib monotherapy. The hazard ratios were 0.57 (95% CI, 0.45–0.73) for TAS102 plus BEV versus TAS102 monotherapy and 0.44 (95% CI, 0.34–0.58) for TAS102 plus BEV versus regorafenib monotherapy. On multivariate analysis, TAS102 plus BEV was independently correlated with better OS and PFS. No unexpected adverse events were observed in any group.
Conclusion:
Our study shows that OS and PFS are longer in patients treated with TAS102 plus BEV than in those treated with TAS102 or regorafenib monotherapy.
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related deaths worldwide. 1 The development of combination chemotherapy regimens that include cytotoxic agents, such as fluoropyrimidine (FU), oxaliplatin (OX), and irinotecan (IRI), and molecular targeted therapies, such as bevacizumab (BEV), ramucirumab, ziv-aflibercept, cetuximab, and panitumumab, has increased the survival of patients with metastatic CRC (mCRC) by around 30 months.2–8 Moreover, recently, the approval of pembrolizumab as a tumor agnostic drug for the treatment of microsatellite instability-high solid tumors and entrectinib for solid tumors with NTRK fusion has contributed to the prolonged prognosis of patients with those gene alterations.9–11
In the CORRECT and RECOURSE phase III trials, the active agents regorafenib and trifluridine/tipiracil hydrochloride (TAS102) significantly improved the overall survival (OS) and progression-free survival (PFS) of patients with chemorefractory mCRC.12,13 Additionally, TAS102 plus BEV was shown to have a promising efficacy in a phase I–II trial (C-TASK-FORCE) 14 and in the recent Danish randomized phase II trial. 15
However, there has been no direct comparative phase III trial to investigate the efficacy of TAS102 plus BEV and either TAS102 or regorafenib monotherapy. Moreover, the effect of TAS102 plus BEV over standard treatment has not been fully demonstrated. Therefore, we evaluated the effects of TAS102 plus BEV, compared with those of TAS102 or regorafenib monotherapy, after standard chemotherapy in patients with mCRC.
Materials and methods
Patients
The present study retrospectively examined the clinical records of patients with mCRC who received later-line chemotherapy comprising TAS102 plus BEV, TAS102 monotherapy, or regorafenib between March 2013 and December 2019 in the National Cancer Center Hospital East (Kashiwa, Chiba, Japan). The main eligibility criteria were: (1) histologically confirmed colorectal adenocarcinoma; (2) no prior treatment with TAS102 and regorafenib; (3) refractory or intolerant to standard chemotherapies [FU, OX, IRI, and anti-epidermal growth factor receptor (EGFR) antibody (for KRAS/NRAS wild-type tumors)]; (4) Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–2; and (5) adequate organ function. The present study was approved by the ethics committee of National Cancer Center Hospital East and was conducted in accordance with the guidelines for biomedical research specified in the Declaration of Helsinki (approved ID: 2017-120). The requirement for informed consent was waived because of the retrospective design of this study.
RAS/BRAFV600E mutation assessment
RAS and BRAFV600E mutation statuses were mainly assessed using the MEBGEN RASKET Kit (Medical & Biological Laboratories, Nagoya, Japan), 16 which could detect 48 types of mutations in codons 12, 13, 59, 61, 117, and 146 of KRAS and NRAS using PCR-reverse sequence-specific oligonucleotide and xMAP technology, or MEBGEN RASKET-B Kit, 17 which could detect a BRAFV600E mutation in addition to 48 types of mutations in KRAS and NRAS. BRAFV600E mutations were assessed using the MEBGEN RASKET-B or MuPACK kits (Medical & Biological Laboratories). 18
Statistical analysis
Efficacy end points included the following: PFS, defined as the time from study treatment initiation to disease progression or death from any cause; OS, defined as the time from study treatment initiation to death from any cause; overall response rate (ORR), defined as the proportion of patients with the best overall complete response (CR) or partial response (PR); and disease control rate (DCR), defined as the proportion of patients with the best overall CR, PR, or stable disease lasting >6 weeks from study treatment initiation. Tumor response was assessed using the Response Evaluation Criteria in Solid Tumors version 1.1. Adverse events were evaluated using the Common Terminology Criteria for Adverse Events version 4.03.
All outcomes were compared between TAS102 plus BEV and TAS102 or regorafenib monotherapy. In addition, we performed subgroup analyses for OS and PFS using the baseline factors. Furthermore, in the patients receiving TAS102 plus BEV or TAS102 monotherapy, we evaluated the prognostic impacts of chemotherapy-induced neutropenia (CIN) within the first cycle (grade 0 versus grade 1 or worse). 14
The following pretreatment clinical data and baseline laboratory values were used as covariates in the analysis: age; sex; ECOG PS; primary tumor site (tumors in the cecum, ascending colon, or transverse colon were classified as right-sided, whereas those in the splenic flexure, descending colon, sigmoid colon, or rectum were classified as left-sided); surgery on the primary tumor; RAS/BRAFV600E status; metastatic tumor site (liver, lung, and peritoneal dissemination); number of metastatic sites; time from the initiation of first-line chemotherapy; and serum carcinoembryonic antigen (CEA). The cutoff value of CEA was set to the median.
Quantitative data were expressed as median and interquartile rang. The Mann–Whitney U test or Kruskal–Wallis test was used to compare continuous variables, and the Fisher’s exact test was used to compare the categorical variables. Survival curves were estimated using the Kaplan–Meier method, and differences between the groups were tested by the log-rank test. Hazard ratios (HRs) were estimated using the Cox proportional hazard model. PFS and OS were analyzed using univariate and multivariate Cox regression analyses. The backward method was conducted for selecting the retained factors (p < 0.2) in the multivariate analysis. The predictive factors for OS and PFS in each group were examined using subgroup analyses.
A 1:1 matching using the propensity score (propensity score-matched dataset) was performed as a sensitivity analysis. The propensity score was calculated with a multivariate logistic regression model, including 12 prognostic variables (Supplemental Material Table 1 online). The patients in the two groups were matched by a difference of propensity score using a 0.05 caliper, equal to 0.2 of the standard deviation of the logit of the propensity score. All p values < 0.05 were considered statistically significant. All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria).
Results
Patients
Of the 497 patients with mCRC, 425 met the inclusion criteria (TAS102 plus BEV, n = 139; TAS102 monotherapy, n = 153; regorafenib, n = 133; Figure 1). The patient characteristics are summarized in Table 1. There was no significant difference between the three groups, except for RAS/BRAF status [RAS/BRAFV600E wild-type: 40.3% (n = 56) versus 35.3% (n = 54) versus 57.1% (n = 76); RAS mutant (MT): 54.7% (n = 76) versus 59.5% (n = 91) versus 38.3% (n = 51); BRAFV600E MT: 3.6% (n = 5) versus 4.6% (n = 7) versus 4.5% (n = 6); p = 0.009], and drug exposure of angiogenesis inhibitors [91.0% (n = 127) versus 90.8% (n = 139) versus 98.5% (n = 131); p = 0.008]. Over 90% of patients had received previous angiogenesis inhibitors, including BEV, ramucirumab, and aflibercept. All patients had measurable lesions.

Patient selection flow diagram.
Patient characteristics of each drug group.

p values were calculated by Fisher’s exact probability test for categorical variables or by Kruskal–Wallis test for continuous variables.
Bevacizumab, ramucirumab, aflibercept.
BEV, bevacizumab; BRAF, v-raf murine sarcoma viral oncogene homolog B1; CEA, carcinoembryonic antigen; ECOG PS, Eastern Cooperative Oncology Group performance status; FU, fluoropyrimidine; IQR, interquartile range; IRI, irinotetcan; MT, mutant; OX, oxaliplatin; RAS, rat sarcoma; WT, wild-type.
Efficacy
The median follow-up at the time of analysis was 25.3 months [95% confidence interval (CI), 19.2–32.2 months]. The median OS and PFS in the overall population were 8.7 months (95% CI, 8.1–9.6 months) and 2.6 months (95% CI, 2.3–3.0 months), respectively. The TAS102 plus BEV group showed significantly longer OS [median 11.5 months (95% CI, 9.9–13.9 months) versus 8.1 months (95% CI, 6.8–9.2 months); HR = 0.67 (95% CI, 0.51–0.88); p = 0.004] and PFS [median 4.4 months (95% CI, 3.7–5.4 months) versus 2.5 months (95% CI, 2.1–3.1 months); HR = 0.57 (95% CI, 0.45–0.73); p < 0.001] compared with the TAS102 monotherapy group. Similarly, TAS102 plus BEV showed significantly longer OS [median 11.5 months (95% CI, 9.9–13.9 months) versus 6.8 months (95% CI, 5.7–8.5 months); HR = 0.71 (95% CI, 0.54–0.94); p = 0.015] and PFS [median 4.4 months (95% CI, 3.7–5.4 months) versus 2.1 months (95% CI, 1.6–2.3 months); HR = 0.44 (95% CI, 0.34–0.58); p < 0.001] compared with the regorafenib monotherapy group (Figure 2). Moreover, the TAS102 plus BEV group showed greater ORR and DCR than the TAS102 monotherapy (ORR: 8.0% versus 1.3%; p = 0.052; and DCR: 64.0% versus 50.0%; p = 0.012) and regorafenib monotherapy groups (ORR: 8.0% versus 0.0%; p = 0.007; and DCR: 64.0% versus 39.5%; p < 0.001) (Table 2).

Kaplan–Meier survival curves for each drug group. (a) The median progression-free survival was significantly longer in the patients receiving TAS102 plus BEV than in those receiving TAS102 monotherapy [4.4 months (95% CI 3.7–5.4) versus 2.5 months (95% CI 2.1–3.1), p < 0.001] or regorafenib [4.4 months (95% CI 3.7–5.4) versus 2.1 months (95% CI 1.6–2.3), p < 0.001]. (b) The median overall survival was significantly longer in the patients receiving TAS102 plus BEV than in those receiving TAS102 monotherapy [11.5 months (95% CI 9.9–13.9) versus 8.1 months (95% CI 6.8–9.2), p = 0.004] or regorafenib [11.5 months (95% CI 9.9–13.9) versus 6.8 months (95% CI 5.7–8.5), p = 0.015].
Overall response.
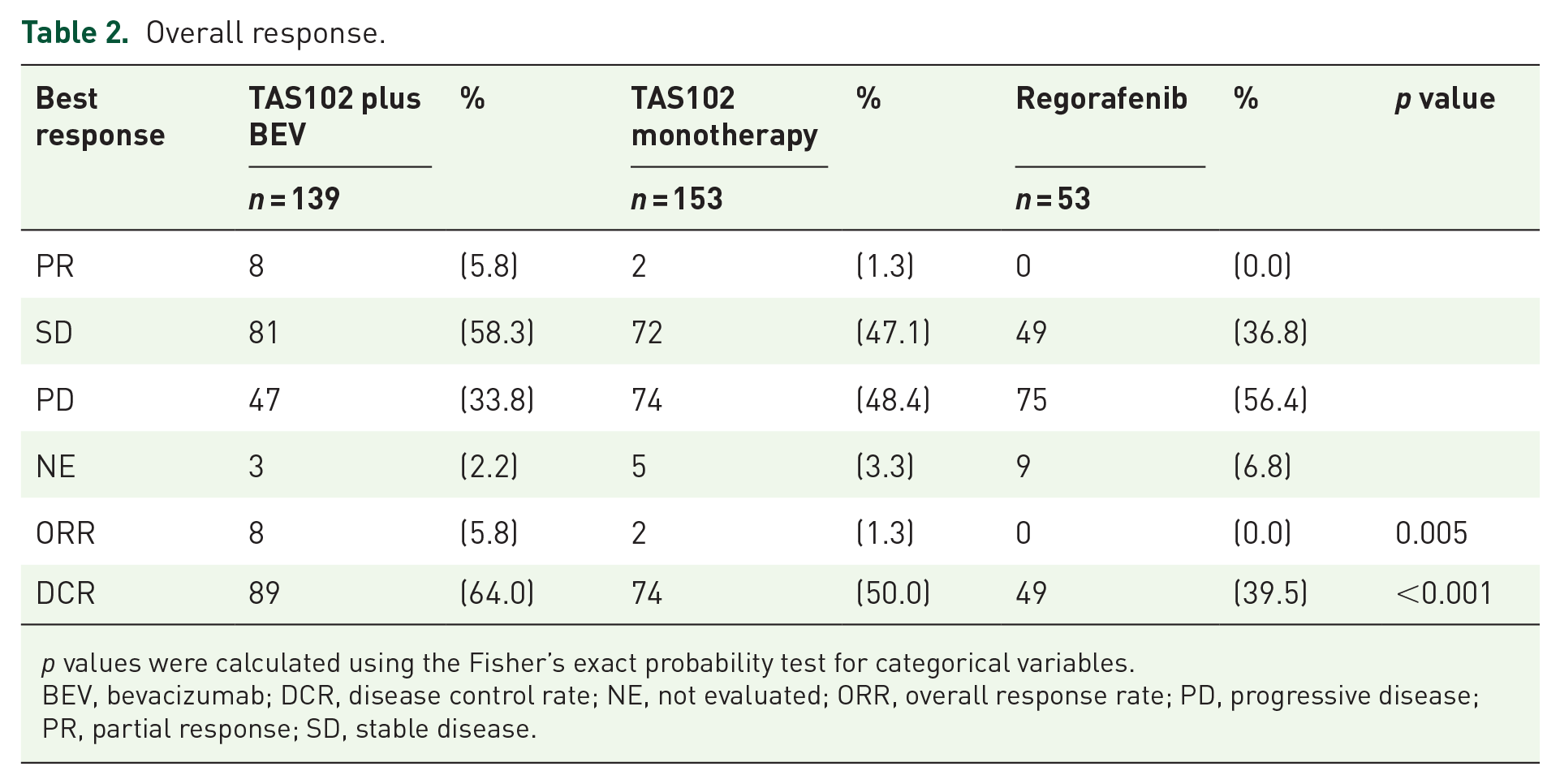
p values were calculated using the Fisher’s exact probability test for categorical variables.
BEV, bevacizumab; DCR, disease control rate; NE, not evaluated; ORR, overall response rate; PD, progressive disease; PR, partial response; SD, stable disease.
The results of univariate and multivariate analyses for OS and PFS are shown in Supplemental Tables S2 (TAS102 plus BEV versus TAS102 monotherapy) and S3 (TAS102 plus BEV versus regorafenib monotherapy). On multivariate analysis, TAS102 plus BEV was significantly associated with longer OS and PFS compared with TAS102 monotherapy (OS: HR = 0.66, 95% CI, 0.49–0.88; p = 0.005; PFS: HR = 0.57, 95% CI, 0.45–0.74; p < 0.001) or regorafenib monotherapy (OS: HR = 0.67, 95% CI, 0.49–0.91; p = 0.010; PFS: HR = 0.45, 95% CI, 0.34–0.60; p < 0.001). In the subgroup analysis for OS and PFS, no significant interactions were observed in any subgroup. Notably, the consistent efficacy of TAS102 plus BEV was observed irrespective of RAS or BRAFV600E mutations [Figure 3(a) to (d)].
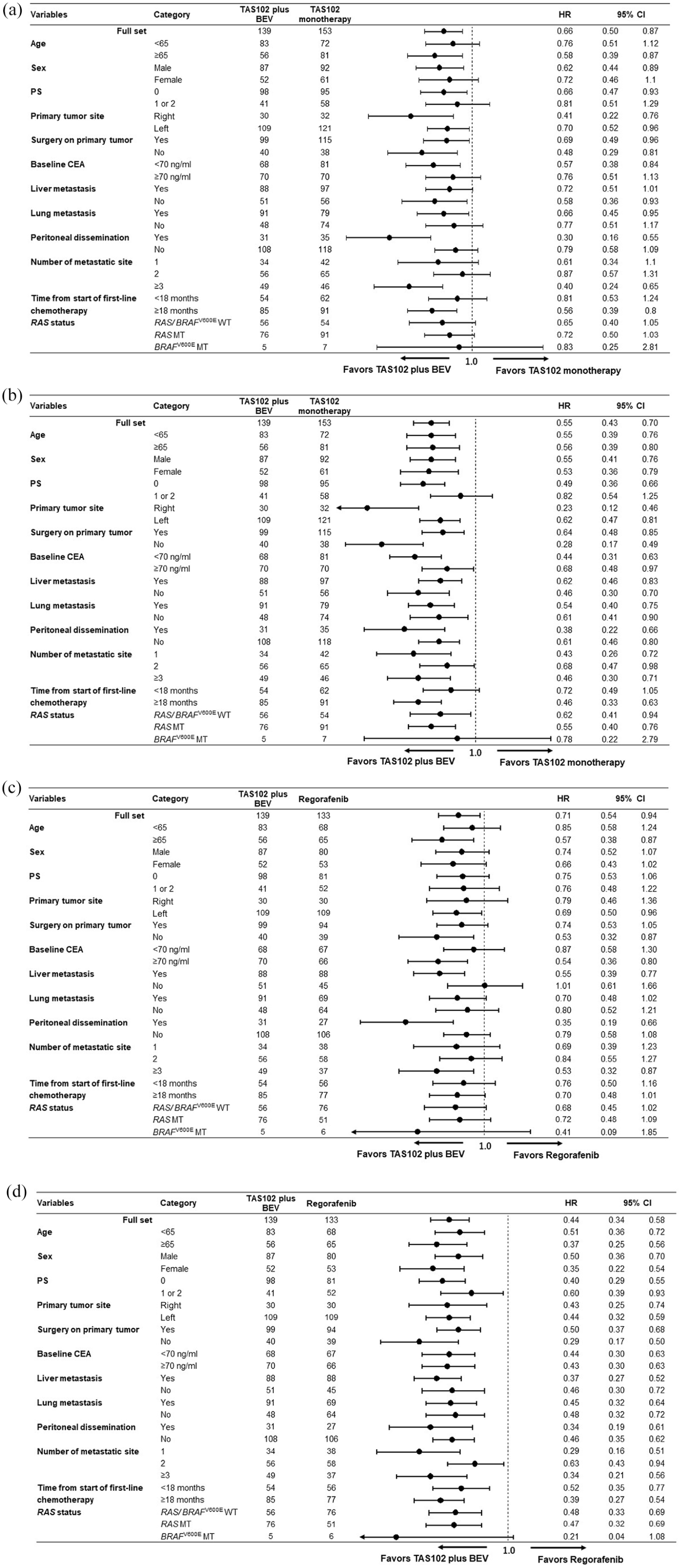
Subgroup analyses for overall survival (OS) and progression-free survival (PFS). (a) OS and (b) PFS for TAS102 plus BEV versus TAS102 monotherapy. (c) OS and (d) PFS for TAS102 plus BEV versus regorafenib.
Prognostic impact of CIN in the TAS102 plus BEV and TAS102 monotherapy groups
In addition, the median OS and PFS between the TAS102 plus BEV and TAS102 monotherapy groups stratified by CIN within the first cycle are shown in Supplemental Figure S1. Between the two groups, CIN of grade 1 or higher within the first cycle was as follows: 59.0% (n = 82) in the TAS102 plus BEV group and 60.8% (n = 93) in the TAS102 monotherapy group. CIN within the first cycle contributed to the longer OS and PFS in both the TAS102 plus BEV and the TAS102 monotherapy groups; TAS102 plus BEV group: [median OS, 12.7 months (95% CI, 10.0–16.8 months) versus 10.3 months (95% CI, 6.7–14.3 months); HR = 0.61 (95% CI, 0.41–0.93); p = 0.021] and [median PFS, 5.3 months (95% CI, 4.0–6.1 months) versus 3.0 months (2.5–5.1 months); HR = 0.60 (95% CI, 0.41–0.87); p = 0.006] and TAS102 monotherapy group: [median OS, 8.9 months (95% CI, 6.8–9.6 months) versus 6.9 months (95% CI, 4.6–8.6 months); HR = 0.70 (95% CI, 0.47–1.04); p = 0.075] and [median PFS, 2.7 months (95% CI, 2.3–3.7 months) versus 2.0 months (1.6–3.0 months); HR = 0.71 (95% CI, 0.50–1.00); p = 0.049]. Moreover, in the patients with CIN, the TAS102 plus BEV group showed significantly longer OS [median, 12.7 months (95% CI, 10.0–16.8 months) versus 8.9 months (95% CI, 6.8–9.6 months); HR = 0.65 (95% CI, 0.45–0.92); p = 0.015] and PFS [median 5.3 months (95% CI, 4.0–6.1 months) versus 2.7 months (95% CI, 2.3–3.7 months); HR = 0.52 (95% CI, 0.38–0.71); p < 0.001] than the TAS102 monotherapy group. Similarly, in the patients without CIN, the TAS102 plus BEV group showed a numerically longer OS [median 10.3 months (95% CI, 6.7–14.3 months) versus 6.9 months (95% CI, 4.6–8.6 months); HR = 0.75 (95% CI, 0.49–1.15); p = 0.182] and significantly longer PFS [median 3.0 months (95% CI, 2.5–5.1 months) versus 2.0 months (95% CI, 1.6–3.0 months); HR = 0.57 (95% CI, 0.40–0.83); p = 0.003] than the TAS102 monotherapy group (Supplemental Figure S1).
Safety and toxicity
In terms of grade ⩾3 adverse events, no significant difference was observed between the TAS102 plus BEV and TAS102 monotherapy groups [69.1% (n = 97) versus 64.7% (n = 103); p = 0.614]. On the other hand, compared with the regorafenib group, the TAS102 plus BEV group had significantly more grade ⩾3 adverse events [69.1% (n = 97) versus 39.1% (n = 52); p < 0.001]. The incidence of grade ⩾3 neutropenia was numerically higher with TAS102 plus BEV than with TAS102 monotherapy, although the difference was not statistically significant (n = 67, 49% versus n = 58, 38%, p = 0.426). Moreover, the incidence of febrile neutropenia was similar between TAS102 plus BEV and TAS102 monotherapy (n = 3, 2% versus n = 8, 5%, p = 0.225), and the number of grade ⩾3 non-hematologic adverse events was similar between TAS102 plus BEV and regorafenib (n = 52, 38% versus n = 43, 32%, p = 0.375). The incidence of grade ⩾3 proteinuria and hypertension with TAS102 plus BEV therapy was significantly higher than that with TAS102 monotherapy (10.7% versus 1.0%, p < 0.001), but was similar to that with regorafenib monotherapy (10.7% versus 17.2%, p = 0.117). The details of the adverse events are listed in Table 3. No significant difference was observed among the three groups in the frequency of emergency hospital admissions [TAS102 plus BEV, n = 22 (15.8%) versus TAS102 monotherapy, n = 26 (17.0%) versus regorafenib, n = 17 (12.8%); p = 0.621]. Further, no treatment-related deaths were recorded.
Frequency of treatment-related grade ⩾3 adverse event.

BEV, bevacizumab.
Subsequent treatment
Among the patients who discontinued the study treatment (TAS102 plus BEV, n = 133; TAS102 monotherapy, n = 152; regorafenib monotherapy, n = 133), the administration of subsequent systemic therapies was significantly more frequent in the TAS102 plus BEV group than in the TAS102 monotherapy group (62.6% versus 42.5%; p < 0.001). In contrast, no significant difference was observed between the TAS102 plus BEV and regorafenib monotherapy groups (62.6% versus 56.4%; p = 0.167). Moreover, no significant difference was found in the proportion of crossover administration of TAS102 and regorafenib between the TAS102 plus BEV and TAS102 monotherapy groups (45.1% versus 36.8%; p = 0.154) and TAS102 plus BEV and regorafenib monotherapy groups (45.1% versus 43.6%; p = 0.903). On the other hand, the percentage of patients who received other systemic treatment, such as reintroduction of OX-based chemotherapy, rechallenge with anti-EGFR antibody, and clinical trials, was higher in the TAS102 plus BEV group than in the TAS102 monotherapy (20.3% versus 4.6%; p = 0.002) and regorafenib monotherapy groups (20.3% versus 8.3%; p = 0.064) (Supplemental Table S4).
Sensitivity analysis
Propensity matching between the TAS102 plus BEV and TAS102 monotherapy groups and the TAS102 plus BEV and regorafenib monotherapy groups revealed that the patients’ characteristics were well balanced. Additionally, as a result, no significant difference in the administration of subsequent systemic therapies was found between the TAS102 plus BEV and TAS102 monotherapy group and the TAS102 plus BEV and regorafenib monotherapy group (Supplemental Tables S5a and S5b). Compared with the TAS102 monotherapy group, the TAS102 plus BEV group showed significantly longer OS [median 11.5 months (95% CI, 9.4–14.3 months) versus 7.6 months (95% CI, 6.2–9.3 months); p = 0.012] and PFS [median 4.2 months (95% CI, 3.0–5.3 months) versus 2.3 months (95% CI, 2.1–3.3 months); p < 0.001] (Supplemental Figure S2). Similarly, compared with the regorafenib monotherapy group, the TAS102 plus BEV group showed significantly longer OS [median 11.3 months (95% CI, 9.6–13.9 months) versus 7.0 months (95% CI, 5.6–8.6 months); p = 0.034] and PFS [median 4.5 months (95% CI, 3.7–5.6 months) versus 2.1 months (95% CI, 1.7–2.5 months); p < 0.001] (Supplemental Figure S2).
Discussion
The phase Ib/II C-TASK-FORCE trial and the randomized phase II Danish trial suggested that TAS102 plus BEV was a promising treatment option. However, based on the results of randomized placebo-controlled phase III trials, the TAS102 and regorafenib monotherapies remain the standard treatment in patients with chemorefractory mCRC.12,13 In terms of the administration sequence, several retrospective studies showed no obvious survival differences between TAS102 first and regorafenib first,19–21 implying that the choice of first treatment should be based on the toxicity profiles and the investigator’s judgment. Our present study showed better outcomes in terms of OS, PFS, ORR, and DCR with TAS102 plus BEV first than with either TAS102 or regorafenib monotherapy first. To the best of our knowledge, this study is the largest cohort study that investigated TAS102 plus BEV therapy for patients with mCRC in a salvage-line setting.
Considering the median OS differences between study drug and the placebo in the CORRECT and RECOURSE trials (1.4 and 1.8 months, respectively), the significant difference in median OS of 3.4 months with HR of 0.67 for TAS102 plus BEV versus TAS102 monotherapy and 4.7 months with HR of 0.71 for TAS102 plus BEV versus regorafenib monotherapy in our study was clinically meaningful in the salvage-line setting. Moreover, using TAS102 plus BEV first was an independent prognostic factor for OS and PFS in multivariate analyses. We used a propensity score matching method to reduce the bias in our retrospective study. Nevertheless, longer PFS and OS were observed in the TAS102 plus BEV group than in the TAS102 or regorafenib monotherapy groups. Although the randomized phase II Danish trial demonstrated the superiority of TAS102 plus BEV over TAS102 monotherapy in PFS and OS, the association of longer PFS and OS with TAS102 plus BEV than with regorafenib has been unknown because of the different characteristics of TAS102 and regorafenib. In our present study, albeit a retrospective analysis, the clinical benefit of TAS102 plus BEV over regorafenib was clearly shown. Furthermore, the consistent efficacy of TAS102 plus BEV therapy compared with the TAS102 or regorafenib monotherapies was demonstrated in all the subgroups in this study. The efficacy of the TAS102 plus BEV therapy for RAS mutant tumors was controversial in the previous studies;14,15 however, our study showed the consistent benefits of TAS102 plus BEV therapy in terms of OS and PFS, irrespective of RAS status.
We also evaluated the relationship between CIN and TAS102 plus BEV or TAS102 monotherapy. Several retrospective studies have suggested that CIN is associated with favorable outcomes in patients with mCRC receiving TAS102.22,23 Importantly, our study demonstrated significantly longer OS and PFS in the TAS102 plus BEV group than in the TAS102 monotherapy group, even in patients with CIN. These findings support the avoidance of dose reduction as long as no safety concerns are raised from an infection standpoint and no neutropenic fever develops in patients undergoing TAS102 plus BEV therapy or TAS102 monotherapy. 24
In terms of safety profile, TAS102 plus BEV was well tolerated in clinical practice setting compared with TAS102 or regorafenib monotherapies on the basis of the side effects. Although there was a numerically higher proportion of grade 3 or 4 neutropenia with TAS102 plus BEV than with TAS102 monotherapy, the frequency of febrile neutropenia or emergency hospital admission did not increase. Our results on the incidence of grade ⩾3 proteinuria and hypertension may be explained by angiogenesis inhibitor-related toxicities. As most patients have been heavily exposed previously to angiogenesis inhibitors as concomitant agents with chemotherapy for mCRC, angiogenesis inhibitor-related toxicities should be monitored carefully in the salvage-line setting.
This study had some limitations that should be considered when interpreting the results. First, this was a non-randomized retrospective study with a limited sample size. The treatment regimen was chosen by each investigator, inducing potential selection bias. Therefore, we performed propensity score matching analysis between TAS102 plus BEV and TAS102 monotherapy group or regorafenib monotherapy group. Propensity score matching demonstrated the consistent efficacy of the TAS102 plus BEV treatment. Second, we could not assess the patients’ quality of life, which is one of the important outcomes in salvage-line setting. Third, all patients enrolled in this study were Japanese. However, in previous phase III trials on the efficacy of TAS102 and regorafenib,12,13,25,26 there were no ethnic differences; therefore, our results may be applicable to all patients, regardless of ethnicity. To validate the results of our study, further prospective investigations in larger cohorts are warranted; an open-label, randomized, phase III study comparing TAS102 plus BEV with TAS102 monotherapy is ongoing in the European Union (EudraCT No. 2020-001976-14).
Conclusion
Our study suggests that TAS102 plus BEV therapy might contribute to a longer OS and PFS, with manageable toxicities, compared with TAS102 or regorafenib monotherapy as the first choice of treatment for patients with chemorefractory mCRC.
Supplemental Material
sj-docx-1-tam-10.1177_17588359211009143 – Supplemental material for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study
Supplemental material, sj-docx-1-tam-10.1177_17588359211009143 for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study by Keigo Chida, Daisuke Kotani, Yoshiaki Nakamura, Akihito Kawazoe, Yasutoshi Kuboki, Kohei Shitara, Takashi Kojima, Hiroya Taniguchi, Jun Watanabe, Itaru Endo and Takayuki Yoshino in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-tif-2-tam-10.1177_17588359211009143 – Supplemental material for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study
Supplemental material, sj-tif-2-tam-10.1177_17588359211009143 for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study by Keigo Chida, Daisuke Kotani, Yoshiaki Nakamura, Akihito Kawazoe, Yasutoshi Kuboki, Kohei Shitara, Takashi Kojima, Hiroya Taniguchi, Jun Watanabe, Itaru Endo and Takayuki Yoshino in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-tif-3-tam-10.1177_17588359211009143 – Supplemental material for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study
Supplemental material, sj-tif-3-tam-10.1177_17588359211009143 for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study by Keigo Chida, Daisuke Kotani, Yoshiaki Nakamura, Akihito Kawazoe, Yasutoshi Kuboki, Kohei Shitara, Takashi Kojima, Hiroya Taniguchi, Jun Watanabe, Itaru Endo and Takayuki Yoshino in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-tif-4-tam-10.1177_17588359211009143 – Supplemental material for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study
Supplemental material, sj-tif-4-tam-10.1177_17588359211009143 for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study by Keigo Chida, Daisuke Kotani, Yoshiaki Nakamura, Akihito Kawazoe, Yasutoshi Kuboki, Kohei Shitara, Takashi Kojima, Hiroya Taniguchi, Jun Watanabe, Itaru Endo and Takayuki Yoshino in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-tif-5-tam-10.1177_17588359211009143 – Supplemental material for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study
Supplemental material, sj-tif-5-tam-10.1177_17588359211009143 for Efficacy and safety of trifluridine/tipiracil plus bevacizumab and trifluridine/tipiracil or regorafenib monotherapy for chemorefractory metastatic colorectal cancer: a retrospective study by Keigo Chida, Daisuke Kotani, Yoshiaki Nakamura, Akihito Kawazoe, Yasutoshi Kuboki, Kohei Shitara, Takashi Kojima, Hiroya Taniguchi, Jun Watanabe, Itaru Endo and Takayuki Yoshino in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
Conflict of interest statement
Daisuke Kotani received honoraria from Takeda, Chugai, Lilly, Merck Serono, Taiho, Ono, and Sysmex. Yoshiaki Nakamura received research grants from Taiho, Chugai, and Genomedia Inc. Akihito Kawazoe received honoraria from Taiho and research grants from Ono, Sumitomo Dainippon, and MSD. Yasutoshi Kuboki received honoraria from Taiho, Sanofi, Bayer, and Ono and research grants from Taiho, Boehringer, AbbVie, GlaxoSmithKline, Chugai, Daiichi Sankyo, and Genmab K.K. Kohei Shitara received honoraria from Novartis, AbbVie, and Yakult and research grants from Astellas, Lilly, Ono, Sumoitomo Dainippon, Daiichi Sankyo, Taiho, Chugai, MSD, and Medi Science. Takashi Kojima received research grants from Ono, MSD, Astellas, BMS, and Merck (H), Ono, MSD, Astellas Amgen, Taiho, and Shionogi. Hiroya Taniguchi received honoraria from Bayer, Sanofi, Takeda, Chugai, Taiho, Lilly, Merck, Yakult Honsha, MBL, Bristol-Myers Squibb, MSD, Novartis, Daiichi Sankyo, Mitsubishi Tanabe, and Nippon Kayaku and research grants from Dainippon Sumitomo, Array BioPharma, MSD, Ono, Daiichi Sankyo, Sysmex, Novartis, and Takeda. Takayuki Yoshino received honoraria from Taiho, Chugai, Lilly, Bayer, and Merck Biopharma and research grants from Taiho, Ono, Amgen, Parexel International, MSD, Chugai, Daiichi Sankyo, Sumitomo Dainippon, and Sanofi. All other authors declare no potential conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.