
Abstract
Cholangiocarcinoma (CCA) is a refractory cancer with limited treatment options and poorly understood molecular mechanisms underlying tumor development. The most effective treatment is surgical resection; however, patients are highly prone to recurrence. Moreover, considering that most patients are diagnosed in advanced stages, treatment options are restricted to palliative care, which results in poor prognosis. Due to the limited effect of chemotherapy and radiotherapy, targeted therapy is becoming a hot topic in the field of biliary cancer treatment. The fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) signaling pathway involves a variety of key biological processes for cell survival, differentiation, and metabolism. Next-generation sequencing data mining has shown that high levels of FGF/FGFR expression are associated with reduced overall survival (OS) in CAA, which indicates that the FGF/FGFR pathway may be an effective target for CAA treatment. This paper reviews the effect of FGF/FGFR signaling on CCA from onset to treatment and highlights the promise of FGF/FGFR signaling pathway inhibitors for targeting CCA.
Keywords
Highlights
Fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) can regulate cell survival, proliferation, and mediate several vital physiological functions such as metabolic homeostasis, neuroendocrine balance, embryonic development, and tissue repair.
Dysregulation of the FGF/FGFR signaling pathway typically occurs through gene amplification, gain-of-function coding mutation, and gene fusion. This consequently affects a series of major biological processes and eventually causes malignancies, including cholangiocarcinoma (CCA).
CCA is a devastating cancer with a frightening 5-year survival rate of approximately 10% and few therapeutic options.
Mutations that alter FGFRs 1–4 are frequently found in CCA, especially FGFR2 fusion and FGFR4 overexpression. Targeted therapies for FGFR signaling pathways in CCA, including small-molecule tyrosine kinase inhibitors (TKIs), FGF ligand traps, and FGFR-targeted monoclonal antibodies, have proven effective and safe in a large number of preclinical and clinical trials.
Targeting FGF/FGFR signaling is a promising treatment approach for CCA. However, to better incorporate FGFR inhibitors into clinical practice, many variables need to be addressed, such as the mechanism underpinning FGFR-inhibitor resistance and possible solutions, the onset of chromosome aberration, and the key to establish effective targeted combinatorial therapies.
Background
Cholangiocarcinoma (CCA) is a highly malignant invasive carcinoma that originates from bile duct epithelial cells; however, the causes of CCA remain unclear. The established risk factors mainly include primary sclerosing cholangitis, bile duct abnormalities, biliary stones within the liver, infection with a liver fluke parasite (a common problem in Asia), exposure to certain chemicals and toxins, hepatitis B and hepatitis C virus infections, and so on.1,2 CCA is cancer with poor-prognosis and low-incidence that is divided into intrahepatic cholangiocarcinoma (iCCA) and extrahepatic cholangiocarcinoma (eCCA) according to anatomical location. 3 Although parts of the same bile duct, eCCA and iCCA were found to express different cell proteins and have different cell morphology, doubling time, chromosome karyotype, and chemotherapy sensitivity in vitro. 4 Furthermore, in contrast with iCCA patients, patients with eCCA usually exhibit jaundice. 5 Their clinical features and biological behaviors also have different characteristics, suggesting that CCAs are heterogeneous in their genotypes and must be studied separately depending on anatomic location. 6
CCA is a devastating cancer with an alarmingly low 5-year survival rate of approximately 10% and few therapeutic options. 7 Therefore, once CCA is suspected, comprehensive clinical examination should be performed to determine its clinical classification and staging. CCAs are relatively resistant to radiotherapy and chemotherapy; even their concurrent use (chemoradiotherapy) can barely improve the survival rate and prolong survival time in partial patients. 8 Currently, the first-line chemotherapy for CCA is limited to 5-fluorouracil alone or its combined use with other chemotherapeutic drugs such as cisplatin. Second-line systemic treatment for advanced CCA typically shows dismal efficacy, with a median progression-free survival (PFS) value of approximately 2.7 months. 9 Surgical resection has been considered the best treatment option for CCA. However, most patients are diagnosed with CCA at an advanced stage and have lost the chance to undergo radical resection (R0 resection). Therefore, it is urgent to find effective targeted therapies for CCA.
FGF/FGFR and its role in tumorigenesis
Greenman et al. reported that more than 1000 somatic mutations found in 274 Mb of DNA maintained consistency with the coding exons of 518 protein kinase genes in 210 diverse human malignancies. 10
Human fibroblast growth factor receptors (FGFRs) – a subfamily of receptor tyrosine kinases – comprise of four family members (FGFR1–4) that interact with 22 ligands (FGF1–14, FGF16–23).11,12 The oncogenic mechanisms of FGF/FGFR signaling are very complicated and not fully understood; FGFs activate FGFRs through paracrine or autocrine mechanisms in cooperation with heparan sulfate proteoglycans. 10 Dysregulation of the FGF/FGFR signaling pathway occurs typically through gene amplification, gain-of-function coding mutation, and gene fusion 13 ;this is usually mediated by fibroblast growth factor receptor substrate 2 (FRS2), mitogen activated protein kinase (MAPK)/extracellular signal-regulated kinase 1/2 (ERK1/2), phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathways, Janus kinase–signal transducer and activator of transcription (JAK–STAT), phospholipase Cγ (PLCγ), ribosomal protein S6 kinase 2 (RSK2) 1,2, and so on.14,15 These processes then lead to intracellular phosphorylation of receptor kinase domains, cascading reactions to intracellular signals, and gene transcription. 16 Many studies have confirmed that the carcinogenicity of FGF/FGFR is a result of acquiring potential mutations that lead to protein-coding and synthesis abnormalities in this pathway, which subsequently affects a series of major biological processes and eventually cause the tumors. However, under physiological conditions, FGF/FGFR can regulate cell survival and proliferation and mediate several vital physiological functions such as metabolic homeostasis, neuroendocrine balance, embryonic development, and tissue repair. 17 In recent years, FGFRs have been also found to stimulate endothelial cell proliferation and promote cancer cell migration, 18 regulate tumor cell proliferation, 19 and activate anti-apoptotic pathways, anti-tumor responses, and angiogenesis.20–22
The FGF/FGFR signaling pathway and CCA
In a study of 4853 tumors, FGFR aberrations were found in 7.1% of cancers, with 66% gene amplification, 26% mutations, and 8% rearrangements, by next-generation sequencing 23 ;notably, these aberrations were distributed as follows: 3.5% FGFR1 (mostly amplification), 1.5% FGFR2, 2.0% FGFR3, and 0.5% FGFR4. Much research has found that FGFR (1–4) and FGF (1–10, 16–19, 22–23) contributes to many cancer-related cancers, such as bladder cancer, lung cancer, breast cancer, gastric cancer, endometrial carcinoma, urothelial carcinoma, bladder cancer, melanoma, hepatocellular carcinoma, multiple myeloma, renal cell carcinoma, prostate cancer, CCA, and so on.12,21,22,24
Previous genomic sequencing studies have revealed that 30–40% of iCCA have actionable mutations, including IDH1/2 (5–20%), FGFR2 fusions (4–16%), ARID1A alterations (7–16%), and BAP1 mutations (1–38%).25,26 Many drugs are being actively developed for the clinical setting, especially those targeting the FGF/FGFR signaling pathway, which is involved in a variety of cancers, including CCA. FGFR2 fusion events were identified in about 13% of iCCA, 7 whereas FGFR4 overexpression was noted in approximately 50% of all CCAs. 27 In addition, FGFR1 and FGFR3 mutations were also detected in CCA. 28
In a previous in vitro study on human CCA specimens, Raggi et al. demonstrated by immunohistochemistry that FGFR1 and FGFR2 were expressed in 30% and 65% of total samples, respectively. 29 Evidently, FGFR1 expression is not consistent in CCA; thus, the of FGFR1 expression in the development of CCA and possible targeted treatment choices need further investigation. The most common FGFR chromosomal aberration in CCA is FGFR2–BICC1 fusion, which is constitutively active and plays a role in the activation of MAPK and PIK3CA/mammalian target of rapamycin (mTOR) pathways. 30 Moreover, a previous study found that 6.6% of iCCAs have the FGFR2 translocation and that FGFR2 amplification portended a better prognosis in 122 Chinese iCCA patients. 31 Overexpression of FGFR2 fusion proteins, generated by genetic translocations, resulted in increased sensitivity to FGFR inhibitors both in vitro and in vivo. 32 Besides, a number of selective FGFR inhibitors, including BGJ398, JNJ-42756493, and AZD4547 have shown meaningful clinical or preclinical activity and manageable toxicities in chemotherapy-refractory CCA.33–35
In addition, high expression of FGF15/19 and FGFR4 is closely associated with bile acid synthesis inhibition, 36 and is also able to slow the progression of inflammatory bile duct diseases. 37 Xu et al. investigated FGFR4 expression in 83 iCCAs and 116 eCCAs by immunohistochemistry, and found that FGFR4 was an independent prognostic factor in iCCAs and perihilar CCAs by multivariate analysis. 38 Moreover, FGFR4 can induce the proliferation, invasion, and epithelial-mesenchymal transition of FGF19+ cell lines in vitro; however, AP24354 – a GFR4 inhibitor – can suppress this phenomenon. Although FGFR4 was found to be associated with poor prognosis in CCA via inducing proliferation, invasion, and suppressing apoptosis, Yoo et al. assessed the expression of 98 genes from 46 iCCAs and found that FGFR4-related genes (FGF19, FGF21, and FGFR4) were significantly associated with better disease-free survival (DFS) in iCCA; these authors even speculated they could be used as biomarkers to define the distinctive molecular phenotype of iCCA. 39
Therefore, targeting FGF/FGFR signaling could be a promising candidate for CCA therapy.
Therapies targeting FGF/FGFR signaling in CCA
Altered FGFR activation results from TKI inhibitor use and triggers intracellular signaling; FGF/FGFR interactions at the extracellular level are associated with monoclonal antibodies and FGF ligand traps. Thus, FGFR inhibitors, which can be divided into FGFR-specific small-molecule TKIs, FGF ligand traps, and FGFR-targeting monoclonal antibodies, are currently being used in preclinical and clinical trials involving patients with advanced malignancies, including CCA.
We used ‘Cholangiocarcinoma/Bile duct cancer/Biliary duct cancer’ and ‘FGFR’ as key words to search for clinical trials on the clinicaltrials.gov site; we then collected detailed information on clinical trials related to FGFR pathway-targeting agents in CCA, as shown in Table 1.
Clinical trials of FGF/FGFR signaling-targeted therapies for CCA.

CCA, cholangiocarcinoma; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; KIT, stem cell factor receptor; PDGF, platelet-derived growth factor; RET, rearranged during transfection; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Small-molecule TKIs of the FGFR
Small-molecule TKIs can either non-selectively inhibit FGFR signaling by competing for ATP binding domains (non-selective inhibitors) or selectively target the kinase domain of FGFRs (selective inhibitors). Considering that FGFRs are a superfamily of receptor tyrosine kinases (RTKs) with catalytic domain structural homology to a certain extent, the first-generation of TKIs was designed to express a multi-kinase activity.40–42 Therefore, first-generation TKIs (such as nintedanib, brivanib, regorafenib, mastinib, E-3810, TSU-68, and BIBF1120) are non-selective inhibitors that can affect VEGFRs, PDGFRs, RET, KIT, and FGFRs. This suggests that, while they extensively target numerous tumorigenic growth factors, they might also indirectly drive tumor progression. Fortunately, however, the second-generation of TKIs (such as AZD4547, dovitinib, ponatinib, BGJ398, and LY2874455) serve as selective FGFR TKIs and are capable of playing targeted therapeutic roles with less toxic effects compared with those of non-selective inhibitors.
BGJ398 is a non-selective inhibitor with potential anti-angiogenic and anti-neoplastic activities; it also participates in the suppression of tumor cell differentiation and proliferation, tumor angiogenesis, and tumor cell survival. A phase II study of BGJ398 in CCAs, with FGFR2 fusions or other FGFR genetic alterations, 33 showed that the overall response rate (ORR) was 14.8%, the disease control rate was 75.4%, and the estimated median PFS was 5.8 months [95% confidence interval (CI) 4.3–7.6 months]. Furthermore, BGJ398 also exhibited manageable toxicities and a high disease control rate (75.4%).
JNJ-42756493 (Erdafitinib), also a non-selective inhibitor, has been granted Breakthrough Therapy Designation by the United States Food and Drug Administration (FDA) for the treatment of urothelial cancer and locally advanced or metastatic urothelial carcinoma in March 2018 and April 2019, respectively. 43 Moreover, a phase I, open-label, multicenter, single-arm, dose-escalation study found that erdafitinib was well-tolerated in Japanese patients with advanced or refractory solid tumors including CCA; currently, a phase II study [ClinicalTrials.gov identifier: NCT02699606] to evaluate the clinical efficacy, safety, and pharmacokinetics of erdafitinib in Asian participants with CCA is ongoing.
TAS-120, which irreversibly inhibits all four FGFR subtypes, can selectively inhibit the growth of human cancer cell lines with FGFR gene abnormalities along with tumor growth in mouse xenograft models. 44 A clinical study reported that the irreversible FGFR inhibitor TAS-120 provides clinical benefit in patients with resistance to BGJ398 or Debio 1347 and overcomes several FGFR2 mutations in iCCA models. 45 A phase II study (ClinicalTrials.gov identifier: NCT02052778) to evaluate the ORR of TAS-120 is underway in approximately 100 iCCA patients with confirmed FGFR2 gene fusions.
ARQ 087 (Derazantinib), a pan-FGFR inhibitor, is currently under clinical investigation [ClinicalTrials.gov identifier: NCT03230318] in patients with FGFR2 fusion-positive inoperable or advanced iCCAs; the results have not yet been announced. However, the latest clinical [ClinicalTrials.gov identifier: NCT01752920] shows that derazantinib has anti-tumor activity and a manageable safety profile in patients with advanced unresectable iCCA with FGFR2 fusion who progressed after chemotherapy; notably, its ORR was 20.7%, the disease control rate was 82.8%, and the estimated median PFS was 5.7 months, although, treatment-related adverse events (AEs) were observed in 93.1% of the patients, including asthenia/fatigue (69.0%), eye toxicity (41.4%), and hyperphosphatemia (75.9%); moreover, AEs with a grade ⩾ 3 occurred in 27.6% patients.
INCB054828 (Pemigatinib) is a novel, selective, oral inhibitor of FGFR 1–3. The recent results of the single-arm, multicohort, phase II FIGHT-202 trial showed that 35·5% CCAs with FGFR2 fusions or rearrangements achieved ORR, included 2.8% complete responses, 33% partial response. 46 But, for the other groups (CCAs with other FGF/FGFR alterations, CCAs with no FGF/FGFR alterations), no responses were observed. The median OS were 21.1 months, 6.7 months and 4.0 months, respectively, in the three groups. These data demonstrate the important role of Pemigatinib in the treatment of CCA with FGFR2 fusions or rearrangements. Now, Pemigatinib has become the first and only FDA-approved targeted-drug for CCA. Results of studies with INCB062079, H3B-6527, and BAY1163877 regarding advanced malignancies, including CCA, are also expected to be released soon.
FGF ligand traps
This approach is based on the development of extracellular “FGF ligand traps” able to bind and isolate FGFs, thereby blocking their interaction with their receptors. For example, FP-1039 is a FGF ligand trap consisting of the extracellular domain of FGF receptor 1 (FGFR1) fused with the Fc region of human immunoglobulin G1 (IgG1). It can selectively block mitogenic FGFs; however, it does not bind hormonal FGFs (FGF19, FGF21, and FGF23), which require co-receptors for binding and downstream signaling.47,48 The phase I dose-finding study of FP-1039 [ClinicalTrials.gov identifier: NCT00687505] and the phase Ib study of FP-1039 in FGFR1-amplified NSCLC [ClinicalTrials.gov identifier: NCT01868022] both showed that FP-1039 was well tolerated even in combination with chemotherapy.49,50 As a result, CCA patients are expected to have even good tolerance to FP-1039; thus, studies on the effects of FP-1039 in CCA treatment should be arranged.
The soluble pattern recognition receptor long pentraxin-3 (PTX3) acts as a multi-FGF inhibitor by binding various FGFs, such as FGF2, FGF6, FGF8b, FGF10, and FGF17. 51 NSC12, a recently discovered small molecule PTX3-derived pan-FGF trap, is able to block FGF2/FGFR interaction and inhibit cell proliferation and tumor growth in human lung cancer cells both in vitro and in an in vivo murine model. 52 This approach seems promising, and, thus, more thought needs to be put into its potential implementation for CCA treatment.
FGFR-targeted monoclonal antibodies
Monoclonal antibodies that specifically target FGFRs or FGFs help in reducing common adverse effects associated with inhibiting multiple FGFR subtypes; for instance, FGFR2 and FGFR3 can be suppressed without compromising adult tissue homeostasis, whereas, since FGFR1 and FGFR4 are closely related, blocking them may lead to metabolic disturbances that have greater health risks.
Vofatamab (B-701) is a monoclonal antibody against mutated FGFR3. It was well-tolerated in a phase Ib/II trial [ClinicalTrials.gov identifier: NCT02401542], and was the first FGF/FGFR inhibitor to qualify for the FDA’s fast-track designation in advanced or metastatic urothelial cell carcinoma. 53
In addition, studies have found that patients with activated/amplified FGFR2 signaling could benefit from GP369 (FGFR2-IIIb-specific monoclonal antibody) and R3Mab (FGFR3-specific monoclonal antibody).11,54
MFGR1877S, a monoclonal antibody against FGFR3, was clinically evaluated [ClinicalTrials.gov identifier: NCT01363024] in patients with advanced solid tumors. The phase I dose-escalation trial included 24 participants, but no results have been announced as of yet. More clinical trial data on FGFR-targeted monoclonal antibodies in CCA therapy are needed to accurately assess their effectiveness.
Disadvantages of current therapies
Resistance to FGFR inhibitions
Multiple nonspecific first-generation FGFR inhibitors are in full swing in CCA clinical trials and have demonstrated non-ideal clinical responses; the median duration of response was found to be only 5–6 months. 55 However, according to research data, second-generation FGFR inhibitors seemingly overcame FGFR-driven lack of specificity in CCA and have improved FGFR activity. 45 Nonetheless, detection of secondary FGFR2 mutations that may confer resistance is imperative in CCA cases.
As we know, tumors are the result of a variety of genetic lesions; clinicians and researchers should not underestimate the ability of tumors to adapt to new stress conditions and resist anti-cancer drugs. Resistance mechanisms to overcome FGFR inhibition are driven by mutation activation or signaling pathway bypassing.
By analyzing pretreatment and post-progression cell-free circulating tumor DNA (cfDNA) in three advanced CCA patients after BGJ398 treatment, Goyal et al. found that FGFR2 point mutations (p.N549H, p.N549K, p.V564F, p.E565A, and p.K659M, p.N549H, p.E565A, p.L617V, and p.K641R) could lead to clinical resistance to FGF/FGFR inhibitions in CCA. 56 However, p.V564F was the only common mutation among the three patients, further highlighting its important role in FGFR2 progression.
Herrera-Abreu et al. used parallel RNA interference genetic screens to show that EGFR plays a role in reducing FGFR-inhibition sensitivity in FGFR3-mutant and FGFR3-translocated cell lines through a negative feedback loop. 57 Besides, activation of members of the EGFR family was also evident in triple-negative breast cancer as a mechanism to resist mutant-cancer gene inhibitors. 58 Moreover, much research has reported that FGFR3 fusions and c-MET may also be involved in resistance to FGFR inhibitors.47,59 Hence, the EGFR family, FGFR3, and c-MET need to be further investigated for possible roles in FGFR-inhibitor resistance in CCA.
Toxicity and side effects of FGFR inhibitors
Unlike VEGFR inhibitors, effective doses of FGFR inhibitors do not induce elevated blood pressure or proteinuria; their most common and severe toxicities include hyperphosphatemia, tissue calcification, and so on.33,60
Increased FGF23 expression is known to be regulated by high serum phosphate and 1,25 (OH)2D levels; FGF23 can down-regulate renal phosphate reabsorption and increase the effect of PTH to up-regulate renal phosphate excretion by reducing the sodium phosphate co-transporters (NaPi-IIa and NaPi-IIc) in proximal tubules, whereas FGFR inhibitors induced the down-regulation of the FGF23-Klotho-FGFR1 complex, thereby leading to hypocalcemia and tissue calcification. 61 Besides, hyperphosphatemia and tissue calcification are correlated with a higher cardiovascular mortality in patients with chronic renal failure, cardiomyocyte and vascular damage, and cardiovascular abnormalities such as impaired contractility, reduced cardiac output (CO), and arrhythmia.62,63
Bétrian et al. have reported two cases where patients developed severe straightening of scalp hair, eyelash trichomegaly, acral desquamation, xerostomia, and heel hyperkeratosis following the use of new selective pan-FGFR inhibitors. 64 We looked up clinical trials related to FGFR inhibitors to find out the corresponding treatment-related side effects. Table 2 shows some common FGFR-inhibitor related adverse events, such as hyperphosphatemia, diarrhea, decreased appetite, fatigue, and liver dysfunction. Severe adverse events were rare; however, it is likely that most of the results came from phase I trials and that the number of clinical trials was too small.
Common toxicities and side effects of FGFR inhibitors.
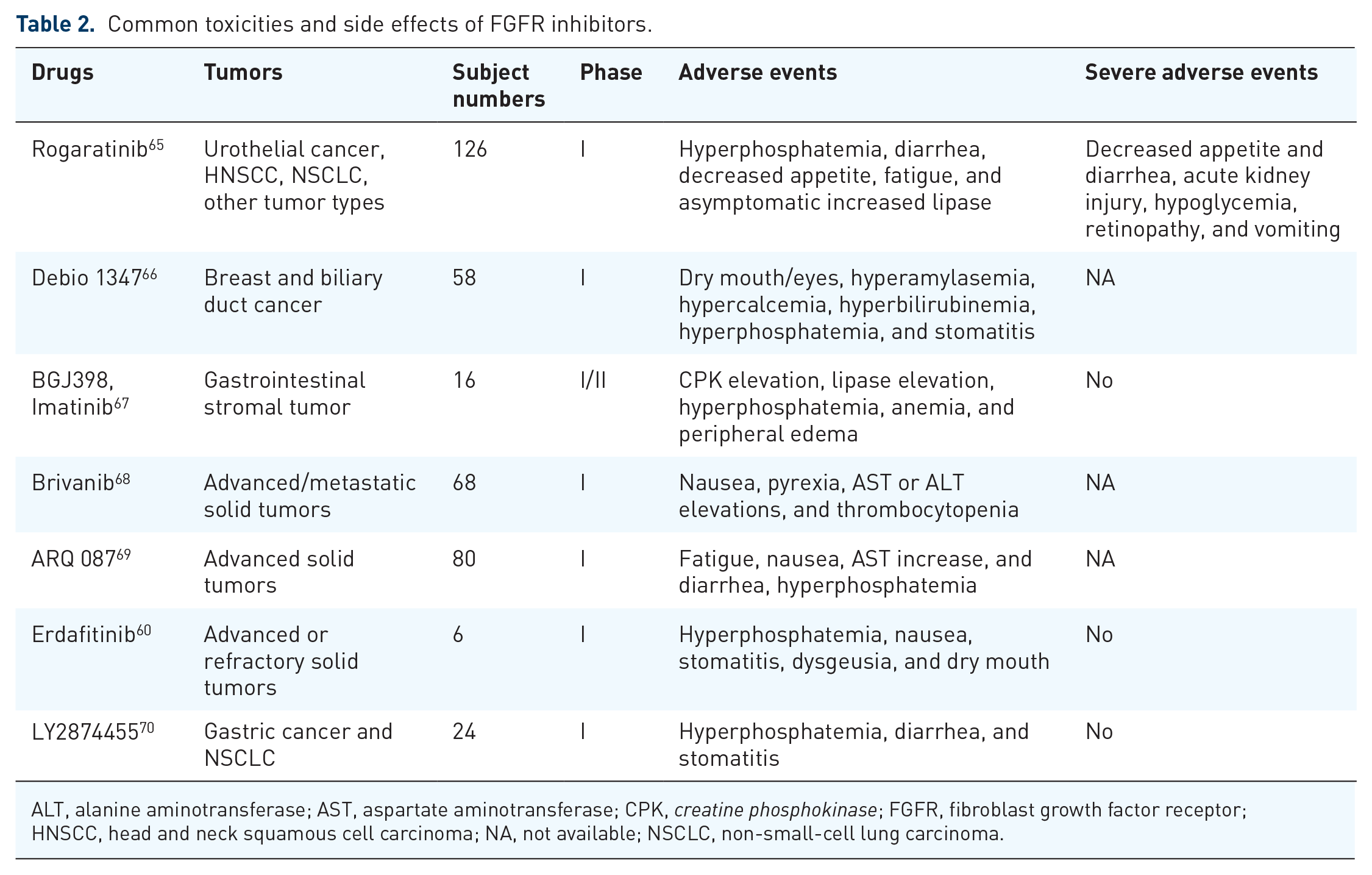
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; FGFR, fibroblast growth factor receptor; HNSCC, head and neck squamous cell carcinoma; NA, not available; NSCLC, non-small-cell lung carcinoma.
In view of the severity of these side effects, the application of FGFR inhibitors may have limits, especially in cancer patients with heart and kidney insufficiency; therefore, developing appropriate side-effect avoidance protocols and/or antagonistic side-effect drugs will be a key point in FGFR-inhibitor promotion.
Conclusion and future perspective
In recent years, genome sequencing has provided a useful approach for cancer diagnosis; FGFR gene mutations were found to be associated with the development of multiple tumors, included CCA. FGF/FGFR inhibitors have been studied extensively as a targeted therapy for CCA. Moreover, the current results have suggested that targeting the FGF/FGFR signaling pathway is promising in CCA.
However, a number of conceptually important questions still remain un-answered; several physiological and pathological processes contribute to the occurrence of CCAs, in which blood and lymphatic angiogenesis processes are also tightly regulated by several key angiogenic factors. The FGF, PDGF, VEGF, and angiopoietin families exert a synergistic effect on tumor blood and lymphatic vessels even though they have different effects on vascular maturation and function, respectively. However, when and how chromosome aberrations occur and the genetic point-of-no-return in CCA remain a mystery. In addition, knowing whether using FGF/VEGF/PDGF/IDH inhibitors in concert (combinatorial or sequential strategies) will provide added benefits for CCA patients, compared with those from their individual uses, is essential information for clinical practice. We believe all of this will facilitate the development of FGFR in the field of CCA.
Footnotes
Acknowledgements
We would like to thank all the scholars whose articles were cited in this paper; without their inspiration and hard work, we would not have been able to complete the final writing of this paper.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The National Natural Science Foundation of China, 81573003. Zhejiang provincial Natural Science Foundation of China-Joint Fund of the Society of Mathematical Medicine, LSY19H160005.