
Abstract
Colorectal cancer (CRC) is a heterogeneous disease representing a therapeutic challenge, which is further complicated by the common occurrence of several molecular alterations that confer resistance to standard chemotherapy and targeted agents. Mechanisms of resistance have been identified at multiple levels in the epidermal growth factor receptor (EGFR) pathway, including mutations in KRAS, NRAS, and BRAFV600E, and in the HER2 and MET receptors. These alterations represent oncogenic drivers that may co-exist in the same tumor with other primary and acquired alterations via a clonal selection process. Other molecular alterations include DNA damage repair mechanisms and rare kinase fusions, potentially offering a rationale for new therapeutic strategies. In recent years, genomic analysis has been expanded by a more complex study of epigenomic, transcriptomic, and microenvironment features. The Consensus Molecular Subtype (CMS) classification describes four CRC subtypes with distinct biological characteristics that show prognostic and potential predictive value in the clinical setting. Here, we review the panorama of actionable targets in CRC, and the developments in more recent molecular tests, such as liquid biopsy analysis, which are increasingly offering clinicians a means of ensuring optimal tailored treatments for patients with metastatic CRC according to their evolving molecular profile and treatment history.
Introduction
Colorectal cancer (CRC) is the third most diagnosed cancer globally, and remains a principal cause of cancer deaths. Although extensive efforts have been made over recent years to optimize tailored therapies in the metastatic setting, the lack of biomarkers for response and the complex heterogeneity of CRC tumor development present largely unsolved challenges. 1 Faeron and Vogelstein described a stepwise model of the adenoma-to-carcinoma sequence, implicating both genetic and epigenetic events in tumor carcinogenesis. 2 The Vogelgram sequence is characterized by multiple steps that involve different oncogenes (e.g. KRAS, NRAS, BRAF, and PIK3CA) and tumor suppressor genes (e.g. APC, TP53, SMAD4, and PTEN) that deregulate key signaling pathways driving disease progression, notably Wnt/β-catenin, the epidermal growth factor receptor (EGFR), downstream mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), and transforming growth factor beta (TGF-β). These alterations are found in the majority of sporadic cancers, which can be divided broadly into two groups 3 : (1) chromosomal instability (CIN) tumors (85%), associated with loss of adenomatous polyposis coli (APC) function and increased copy number alterations (CNA) related to TP53 mutations; and (2) tumors with microsatellite instability (MSI; 15%), which are hypermutated as a result of a defective DNA mismatch repair (MMR) system, with a clear molecular origin (MLH1, MSH2, MSH6, or PMS2 inactivation) arising from germline or somatic mutations.
Today, RAS remains the only validated biomarker of resistance to anti-EGFR treatments, but evidence from preclinical and clinical studies is converging to highlight a role for other biomarkers acting as molecular drivers of primary and acquired resistance in CRC. The use of innovative and evolving techniques such as next-generation sequencing (NGS) assays and targeted gene expression profiling to assess multiple gene alterations and signatures over time, have great potential to guide drug development in the context of molecularly defined subtypes. This review discusses the latest progress in biomarker discovery and how transcriptomic and microenvironment profiling may be merged with genomic, epigenomic, immune-stromal notions to stratify patients, with an overlying aim of ensuring the optimal treatment option in the metastatic CRC (mCRC) setting.
Oncogenic biomarkers
RAS mutations in tissue and plasma
Despite extensive efforts, the RAS oncogene, mutated in more than 50% of mCRC cases, is still the unique validated biomarker of resistance to anti-EGFR monoclonal antibodies (mAbs), but remains an elusive target in this setting. Preliminary data from a phase I trial exploring AMG510 – a KRAS-G12C selective inhibitor – in cohorts of patients with metastatic lung cancer or CRC, demonstrate a favorable safety profile and efficacy (50% of lung cancer patients achieved a partial response, and 10 of 19 patients with mCRC had stable disease at the first data cut-off).4,5 These results clearly need to be confirmed in larger randomized clinical trials.
The introduction of innovative molecular tests has resulted in major steps forward in our understanding of the RAS pathway and its clinical implications, leading to the broad stratification of CRC patients applied clinically today, of KRAS and NRAS mutant versus wild type (WT) tumors (Table 1). RAS mutated cancers represent a group of diseases whose common feature is that they do not respond to anti-EGFR drugs, with the main treatment guidelines now reserving the use of these therapies for the RAS WT population. However, retrospective analyses from phase III randomized clinical trials suggest that patients with very low tumoral RAS mutant allele fractions (MAFs) may benefit from cetuximab and panitumumab, given their behavior resembling that of RAS WT tumors. In particular, in the phase III CRYSTAL trial, a better outcome was derived from the addition of cetuximab to a FOLFIRI first-line regimen in mCRC patients with RAS MAFs < 5%. 6 The results were in line with several studies reporting 1–5% cutoffs for the presence of KRAS subclones for anti-EGFR treatment benefit by digital PCR (dPCR) analysis in patients treated with cetuximab or panitumumab, although these retrospective data should be validated in larger prospective trials.7,8 The ULTRA phase II trial provided evidence of higher progression-free survival (PFS), overall survival (OS), and overall response rate (ORR) for patients without mutations in KRAS, NRAS, BRAF, and PIK3CA exon 20 detected by a dPCR analysis using a 5% MAF threshold, compared with lower cut-offs, suggesting that higher sensitivity could impair patient selection. 9 The cut-off discrepancies between the different publications are likely explained by heterogeneity of liquid biopsies analyses and patient populations – an issue that requires further clarification.10,11 Circulating tumor DNA (ctDNA) studies have provided major insights into the identification of genomic alterations such as RAS and other molecular drivers, in cases of acquired resistance to anti-EGFR therapies, and in tumors initially diagnosed as WT. 12 In most cases, different alterations coexist in the same tumor, such as KRAS, NRAS, and BRAF mutations and MET or ERBB2 amplifications, because of the presence of minor subclones pre-existing in the tumor that are detected during treatment, conferring resistance to anti-EGFR drugs. Another mechanism of resistance seen in plasma after the detection of RAS mutated clones is the presence of EGFR extracellular domain (ECD) mutations that impede the binding of mAbs to the EGFR receptor in approximately one-third of cases. 13
Principal first line phase II/III clinical trials with cetuximab and panitumumab in RAS WT population.

5-FU, 5-fluorouracil; Bev, bevacizumab; Cet, cetuximab; LFA, leucovorin, maint, maintenance, mOS, median overall survival; mPFS, median progression free survival; ORR, overall response rate; Pan, panitumumab; WT, wild type.
The PROSPECT-C trial showed how the longitudinal analysis of liquid biopsies integrated with a mathematical modeling of tumor evolution allows to a complete monitor of genetic variations and an early detection of progression, providing innovative personalized treatment strategies. 14 Evolving ctDNA analyses at different time points have revealed how clones rapidly decline following drug withdrawal, potentially restoring sensitivity to anti-EGFR agents. 12 Mutation kinetics are characterized by an inverse relationship between the clones’ decay and the time since anti-EGFR withdrawal, with a median half-life of 4.4 months. The half-life data are in line with previous studies of patients who responded to re-challenge with cetuximab after a treatment break of 4.6 months, suggesting that, if an anti-EGFR re-treatment were taken into account, waiting two half-lives should be considered, and this approach is currently being investigated in many open studies. 15
Further retrospective analyses with anti-EGFR re-challenge in third-line were performed in patients who had received first-line cetuximab plus irinotecan-based therapy, and a subsequent line without anti-EGFR drugs (Table 2). Outcomes were better in patients who benefited in the first-line setting with cetuximab, and, notably, in those with a longer interval between the last dose of cetuximab and progression on second-line therapy.16–18 Moreover, the prospective phase II Cricket trial demonstrated an ORR of 21.5% with cetuximab-based re-challenge treatment, with higher PFS and OS when patients selected by ctDNA analysis did not present RAS mutation in plasma. 19 This, and the previous results, strongly support the value of ctDNA-based selection of patients for the sequential use of cetuximab or panitumumab in refractory patients. The re-challenge strategy has also been investigated with novel anti-EGFR compounds. In a randomized phase II trial with Sym004, a mixture of two EGFR mAbs, patients with plasma ctDNA negative for RAS, BRAFV600E, and ECD activating mutations at the time of treatment initiation had an increased median OS by 5.5 months compared with standard-of-care rescue therapies. 20 These promising results in a molecularly selected population support the design of pivotal ctDNA-guided clinical trials in EGFR inhibitor refractory mCRC.
Past and ongoing anti-EGFR rechallenge clinical trials.

EGFR, epidermal growth factor receptor; mCRC, metastatic colorectal cancer; mPFS, median progression free survival; ORR, overall response rate.
BRAF mutations
BRAFV600E mutations occur in 8–10% of mCRC and are frequently associated with MSI and RAS WT tumors. This genetic alteration is associated with worse prognosis, and could predict a poorer response to anti-EGFR treatment.21,22 Based on the exciting results achieved in melanoma patients, a number of trials investigated the use of BRAF inhibitors as single agents, or in combination with MEK inhibition, in the mCRC setting but have not met with the same success (Table 3). The explanation may be related to suboptimal blockade of the MAPK pathway, the main convergent escape mechanism of all genomic alterations that result from acquired resistance to these agents in CRC. CRC tumor cells rely on EGFR activation as a feedback mechanism upon BRAF inhibitor exposure, with PI3K-mediated sustained MAPK signaling and activation. 23 In clinical trials in mCRC, combinations of BRAF inhibitors plus EGFR and/or MEK inhibitors achieved response rates of 15% to 25% (Table 3). A randomized phase II trial recently demonstrated a 2.4-month improvement in median PFS with the addition of vemurafenib to the control regimen of irinotecan plus cetuximab. 24 The BEACON trial evaluated encorafenib and cetuximab +/– binimetinib (triplet or doublet combination) versus investigator’s choice of irinotecan or FOLFIRI + cetuximab (control) in BRAFV600E mutated refractory mCRC (Figure 1). Patients treated with both double and triple combinations achieved comparable mOS of 9.3 months, compared with 5.9 months in the control arm. The confirmed ORR was 27% with the triple combination of encorafenib, binimetinib, and cetuximab, and 20% with the encorafenib plus cetuximab doublet, with an acceptable toxicity profile.25,26 Of note, double and triple combinations showed prolonged maintenance of quality of life (QoL), reducing the risk of QoL deterioration by more than 40%. 27 These encouraging data show for the first time that patients with BRAF V600E CRC could benefit from a triple combination of targeted therapy, avoiding the need for chemotherapy and its toxic side effects, changing the course of this very aggressive disease.
Clinical trials exploring anti BRAF-EGFR combinations in mCRC.

EGFR, epidermal growth factor receptor; mCRC, metastatic colorectal cancer; mPFS, median progression free survival; mOS, median overall survival; ORR, overall response rate.

Triple anti-EGFR/BRAF/MEK combination in the BEACON trial.
Special reference needs to be made to BRAFnon-V600E mutations, most importantly BRAFD594/G596 associated with impaired kinase activity. 28 The frequency of BRAFnon-V600E mutations is almost 2% by NGS detection and in some cases it coexists with RAS mutations. These mutations do not seem to affect prognosis of patients with mCRC, with their maintained sensitivity to anti-EGFR drugs; nonetheless, prospective randomized clinical trials are needed.29,30
ERBB2 and MET amplifications and kinase fusions
HER2 has been proposed as a target in CRC following studies of RAS/BRAF WT and cetuximab-resistant CRC xenograft models. Molecular alterations in this receptor are a recognized mechanism of primary resistance to anti-EGFR agents in a quadruple WT population (KRAS, NRAS, BRAF, and PIK3CA WT), but also a predictive marker of response to anti-HER2 treatment in mCRC. 31 HER2 overexpression has been observed in 2–3% of genetically unselected population, reaching 5% in left-sided RAS and BRAF WT CRC. 32 HER2 overexpression is determined using tissue-based techniques such as immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), and tissue NGS, as well as less invasive methods such as blood-NGS and ctDNA analyses.
To examine the value of HER2 as a positive predictive biomarker, a proof of concept study using CRC liver metastates xenograft models was carried out with different HER2-targeted therapies, alone or in combination. Whereas the use of HER2 tyrosine kinase inhibitors or anti-HER 2 mAbs as single agents was not effective, the combination of lapatinib plus trastuzumab resulted in activity in the subset of cetuximab-resistant, quadruple WT ERBB2-amplified mCRC xenopatients, with achievable implications in the clinical setting. 32 Based on these preclinical results, the phase II HERACLES trial was launched to assess the ORR of trastuzumab combined with lapatinib (cohort A) or pertuzumab (cohort B) in KRAS exon 2 (codons 12 and 13) WT and HER2-overexpressed mCRC patients resistant to standard therapies, including anti-EGFRs. 33 A 30% ORR with 38 weeks median duration of response was seen for the lapatinib-trastuzumab combination. Notably, responses were significantly more common in tumors with a high ERBR2 gene copy number, and PFS was longer in this population. A plasma copy number (pCN) analysis of 29 patients suggested a pCN threshold predictive of anti-HER2 response, showing an ORR of 35% and a PFS of 4.5 months in patients with pCN > 25.82. 34 Double HER2 targeting with pertuzumab and trastuzumab was explored in the phase II basket study MyPathway, which gave an ORR of 38% in ERBB2-amplified mCRC. 35
The most recent data come from three ongoing clinical trials (Figure 2). In the MOUNTAINER trial, the combination of tucatinib (an anti-HER2 small inhibitor) plus trastuzumab provided the best results to date in patients with HER2-amplified CRC, reporting an ORR of 55%. 36 Moreover, the GOZILA sub-trial confirmed the efficacy of double blockade of the HER2 receptor with trastuzumab plus pertuzumab in patients with HER2-positive tumors selected by tissue and ctDNA analysis, with an ORR of 35%, with both methods able to detect HER2 amplification in CRC. 37 On the other hand, the above-mentioned HERACLES cohort B trial, in which patients progressing in the HERACLES cohort A trial received the antibody-drug conjugate trastuzumab-emtansine (T-DM1) plus pertuzumab combination, did not reach the primary endpoint, with T-DM1 not considered a promising drug in this setting, possibly linked to the poor response to microtubule inhibitors (emtansine) in CRC. However, a 70% disease control and a favorable tolerability profile, similar to those of the HERACLES A trial, make this drug an option in patients with low tumor burden. 38 These emerging data are being validated in multiple ongoing trials with different drug combinations, but a phase III trial has not yet been initiated (Table 4).

Recent anti-HER2 strategies in mCRC. MET tyrosine kinase receptor activation and overexpression have been investigated as a potential mechanism of both primary and acquired resistance to anti-EGFR treatment and BRAF inhibitor combinations in BRAFV600E mutant CRC. 39 Data from in vitro and in vivo studies have shown that CRC xenograft models with MET amplification do not respond to anti-EGFR antibodies. 40 MET amplifications have been found in only a small percentage (1–2%) of the CRC population upfront, but a study found that using NGS liquid biopsies in an anti-EGFR refractory setting the prevalence of MET amplification could rise to 20%. 41 Preliminary studies with MET inhibitors such as SYM015 suggest the resistance could be overcome, with a clear implication for ongoing clinical trials currently exploring these agents. 42
Recent and ongoing trials with anti-HER2 combinations in mCRC.

FISH, fluorescent in situ hybridization; IHC, immunohistochemistry; mCRC, metastatic colorectal cancer mOS, median overall survival; mPFS, median progression free survival; NGS, next generation sequencing; ORR, overall response rate; T-DM1, trastuzumab emtansine.
ALK, ROS1, NTRK1, NTRK2, NTRK3, and RET fusions were identified to be targetable in mCRC, occurring in only ~1.5% of all CRCs. 43 Preclinical models with rearrangements involving these genes are particularly sensitive to kinase inhibitors and patients with fusion-positive tumors present impressive responses to the pan ALK/ROS1/TRK inhibitor entrectinib. 44 Exceptional responses were also obtained in three of four mCRC patients treated in the basket trial with larotrectinib, as well as in two patients with either an ALK or an NTRK1 rearrangement treated with entrectinib.45,46 Rearrangements were enriched in the older population, RAS and BRAF WT, MSI, and right-sided colon cancers, indicating a targeted approach for these patients, with an implementation of fusion analysis in the standard molecular screening.47,48 Of note, in the last 2 years, NTRK inhibitors have been approved by regulatory agencies for all solid tumors with NTRK1, NTRK2, and NTRK3 fusion genes, and for NSCLC with ROS fusion genes.
CMS: a multilevel CRC classification
The research community has attempted to classify molecular subtypes of CRC with similar molecular alterations. Six independent transcriptomic classifications have been proposed by the international Colorectal Cancer Subtyping Consortium and four consensus molecular subtypes (CMS) have been merged as a result of the inter-connectivity of 27 subtypes.
The CMS classification identified subtypes with convergent pathway features: CMS1 tumors (MSI immune), which are hypermutated, MSI, immune-infiltrated, enriched for BRAFV600E; CMS2 (canonical) which are epithelial; CIN, immune desert, driven by the Wnt pathway and MYC activation, with marked EGFR signaling dependence; CMS3 (metabolic) which are epithelial, with metabolic deregulation, mixed chromosomal and MSI, and ubiquitous MAPK pathway alterations; and CMS4 tumors (mesenchymal), which are CIN, with activated TGF-β and vascular endothelial growth factor receptor (VEGFR) pathways (Figure 3). 49
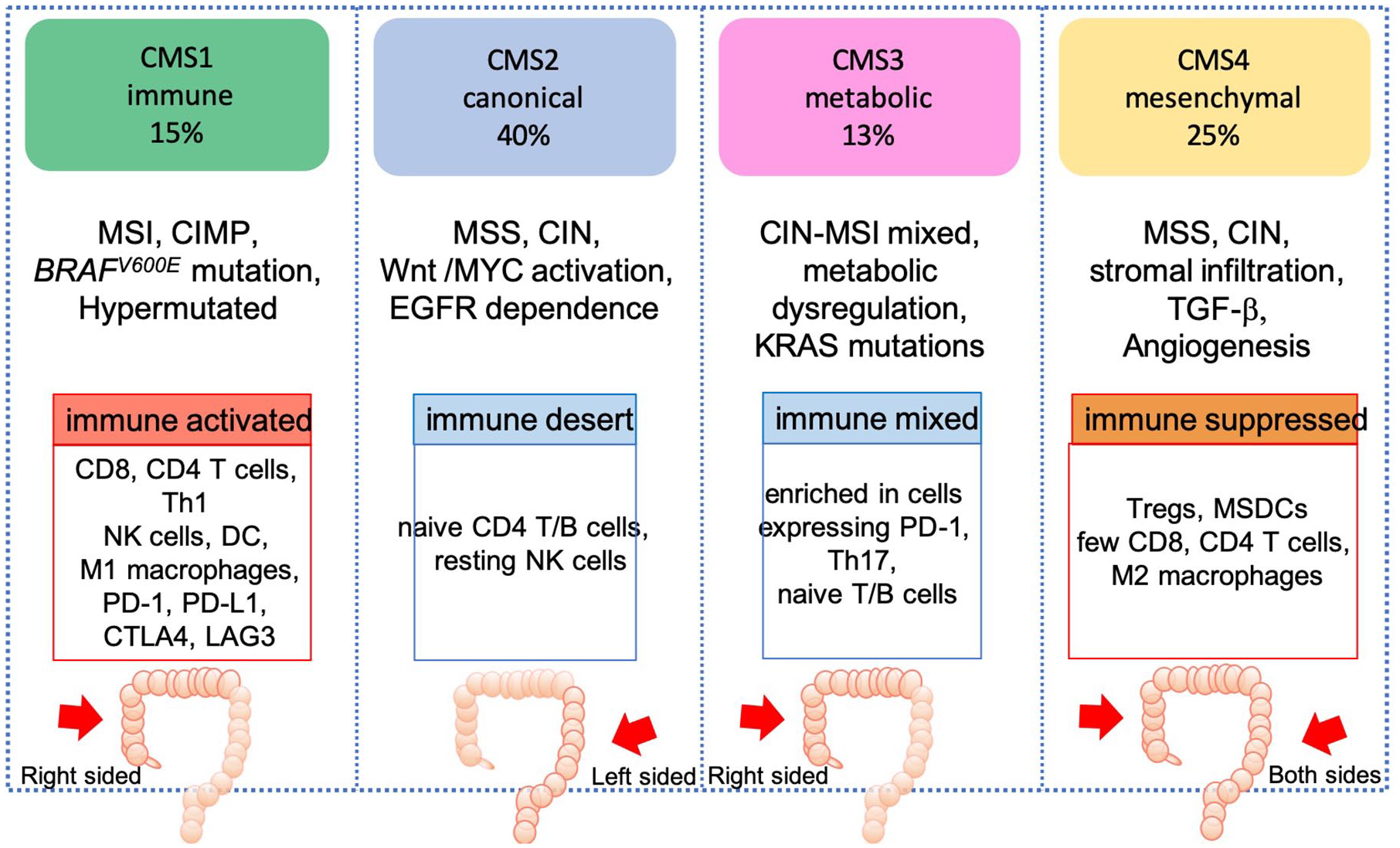
CMS classification.
The biological associations of CMS subtypes with clinical outcomes have been extensively validated in retrospective analyses of clinical trials and the prognostic role attributed to CMS has been confirmed in the metastatic setting. CMS2 canonical tumors have the best prognosis in the metastatic setting, whereas CMS1 MSI immune tumors present an increased risk of progression and death following standard chemotherapy and targeted agents.49,50 Moreover, CMS1 MSI or POLE ultramutant CRC are known to respond to immunotherapy, whereas CMS4 mesenchymal tumors, which have a poorer prognosis compared with other subtypes, show activation of immunosuppressive molecules by the tumor microenvironment.
Although the aim of the CMS classification is to define a “consensus” for CRC transcriptomic subtyping, translation into the clinic is complicated by the complex tumor heterogeneity associated with CRC, and, while its prognostic value has been established for certain settings, its predictive value remains to be confirmed.
Tumor location differs across the CMS subtypes, with CMS1 more enriched in right-side CRC tumors, whereas the “canonical” CMS2 is predominantly found in left-sided, EGFR-dependent, colon cancers. 51 Moreover, differences have also been associated with the transition from adenoma to the latest stages of the disease. In the metastatic setting there is an enrichment in CMS4 mesenchymal tumors, with fewer cases of CMS1 and CMS3, compared with stage II and III CRC (Figure 3). 52
A recent study showed that 57% of primary tumor samples of patients enrolled in the PETACC-8 adjuvant trial demonstrated intratumor heterogeneity in CMS classification, with the presence of two or three different sub-clonal CMS populations in the same sample.53,54 This strong heterogeneity could be explained by gene expression variations between different regions of the tumor, associated with tumor microenvironment components. Changes in the tumor microenvironment are linked to prognosis but can also result in inaccurate CMS subtyping. CMS subclassification is affected by changes from the tumor core to the invasion front, linked mainly to immune-stromal content. In the future, the use of transcriptomic signatures based on cancer cell-intrinsic gene expression may overcome the confounding effect of the tumor microenvironment on patient classification. 55
The prognostic value of CMS subgroups has been extensively studied over the last few years, mainly in the early-stage setting, identifying a worse outcome for patients with CMS4 mesenchymal tumors. Multivariable models suggest that the prognostic value of CMS groups is largely explained by infiltration patterns of stromal cells and cytotoxic T cells. 56
Regarding the predictive value of the transcriptomic classification for adjuvant chemotherapy, a recent study aimed to confirm previous results showing an oxaliplatin benefit in CMS2 patients treated in the C-07 adjuvant study, by retrospectively analyzing patients treated in the MOSAIC trial. However, the results were not significant in terms of benefit with oxaliplatin in the CMS2 population. 57
In the metastatic setting, retrospective analyses of large clinical trials have been conducted to investigate an association between CMS groups and activity of cetuximab or bevacizumab. Both the CALGB-80405 and FIRE3 trials reported poor survival for patients with CMS1 tumors, intermediate survival for CMS4, and good outcomes associated with CMS2 across the two treatment arms. 58
In CALGB-80405 trial, 392 samples were analyzed by Nanostring, finding a clear OS advantage for CMS1 tumors treated with bevacizumab (p < 0.001) compared with cetuximab, whereas CMS2 tumors were characterized by longer OS when treated with cetuximab upfront. These results have been confirmed by a validated metanalysis of six clinical datasets, with a benefit in left-sided colon tumors (largely CMS2) treated with anti-EGFR mAbs. 50 The worse outcome in CMS1 tumors treated with cetuximab in the phase III CALGB-80405 trial is dependent on the presence of well-known mechanisms of resistance to anti-EGFR drugs such as MSI and BRAFV600E mutations. These results are partially discordant with the FIRE3 study, in which the benefit from cetuximab in terms of PFS and OS is reported only in the CMS4 subgroup. 59 There are major differences that could explain the discrepancies between these two studies and the lack of reproducible associations between CMS and benefit from specific therapies, including differences in patient cohorts (RAS WT, high prevalence in left-sidedness in the FIRE3 trial versus KRAS WT, high prevalence of right-sidedness, MSI tumors in the CALGB-80405 trial), chemotherapy backbone (irinotecan versus oxaliplatin), the tumor microenvironment, and the biology and heterogeneity of CRC. 54
Further efforts are needed to refine the CMS classification, integrating DNA signatures, a deeper “functional” characterization of cancer cells, and the tumor microenvironment, which could improve the knowledge of spatial and temporal intra-tumor heterogeneity that impair the association between CMS classification and treatment benefit.
Oncogenic signatures
MSI and “MSI-like”
The presence of MSI varies with tumor stage, being reported in 20% of cases in stage II, 12% in stage III, and 5% in stage IV disease. Most MSI CRC tumors are sporadic and associated with MLH1 loss via promoter methylation/biallelic somatic genomic alterations, whereas 3% harbor germline mutations in MLH1, MSH2, MSH6, and PMS2 genes, as in the case of Lynch Syndrome. 1
Various diagnostic tests can be used to detect MSI tumors, such as IHC panels of MMR proteins and polymerase-chain reaction (PCR)-based assays for microsatellite loci, and NGS panels that measure the tumor mutational burden (TMB) or unstable microsatellite repeats compared with a control, with a high level of concordance.60,61 MSI tumors show a high TMB due to the extensive number of events that occur during DNA replication, including deletions, insertions, and frameshift mutations. They generate neoantigens that are presented by major histocompatibility complexes (MHC) to T-cells and recognized as foreign. This phenomenon leads to high CD8-positive cytotoxic T lymphocyte infiltration and consequent INFγ production, a hallmark of MSI tumors, and to the expression of immune checkpoint proteins such as programmed cell death 1 receptor (PD1), programmed death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), among others, making these tumors particularly responsive to immunotherapy. 62
Immune checkpoint inhibitors (ICI) have been evaluated in patients with MSI mCRC (Tables 5 and 6). The KEYNOTE 016 basket trial tested the efficacy of the anti-PD1 inhibitor pembrolizumab in patients with treatment-refractory progressive metastatic MSI tumors. An ORR of 52% was reported, with a 2-year PFS of 59%, a 2-year OS of 72%, and a grade 3–4 treatment-related adverse event rate of 17%. 63 The CheckMate 142 non-randomized phase II clinical trial reported an ORR of 31% in a refractory mCRC population with the anti-PD1 mAb nivolumab as single agent, whereas the combination of nivolumab with the anti-CTLA4 mAb ipilimumab reached a 55% ORR, with a 1-year PFS rate of 71% and a grade 3–4 adverse event rate of 32%.64,65 Recently, the results of nivolumab plus low-dose ipilimumab in the first-line setting were presented, giving a 60% ORR, and 1-year PFS and OS rates of 77% and 83%, respectively, and a grade 3–4 adverse events rate of 16%. 66
Recent clinical trials with immune checkpoint inhibitors in mCRC.

mCRC, metastatic colorectal cancer; mOS, median overall survival; mPFS, median progression free survival; MSI, microsatellite instability; MSS, microsatellite stability; ORR, overall response rate.
Selection of ongoing trials with immune checkpoint inhibitors in mCRC.

Bev, bevacizumab; mCRC, metastatic colorectal cancer; MSI, microsatellite instability; MSS, microsatellite stability; RT, radiotherapy.
Given these impressive results, even with the lack of randomized phase III clinical trials of ICIs in mCRC, the FDA approved pembrolizumab and nivolumab in patients who have progressed on irinotecan and oxaliplatin-based treatments. Phase III randomized trials in the first-line setting are ongoing, including the KEYNOTE 177 trial, evaluating pembrolizumab versus standard-of-care in untreated MSI mCRC tumors, the COM-MITT trial, exploring atezolizumab as a single agent or in combination with FOLFOX plus bevacizumab versus FOLFOX bevacizumab alone in the first-line setting (Table 6). 67
Although immunotherapy shows activity in the MSI population, not all MSI patients respond to the treatment. No biomarkers of immune response have been identified to date, possibly because PD-L1 expression, BRAF mutation status or the genetic basis for MMR deficiency are not related to ICI efficacy.63,64 TMB has been established as a biomarker of response to ICIs in several tumor types, particularly in MSI cancers.68,69 Emerging data in a cohort of 22 patients with MSI mCRC pointed to the predictive role of TMB for PD-1/L1 inhibitor response, suggesting an optimal cut-point in the range of 37–41 mutations/Mb. 70 These promising results require further validation in larger prospective analyses, to identify those patients with high TMB to be selected for an upfront immunotherapy strategy.
Other immune responders
It is not only MSI tumors that respond to immunotherapy strategies. The POLE-mutation confers the highest mutation rates, present in 1–2% of MSS CRC tumors, with tumor-infiltrating lymphocyte (TILs) infiltration and increased cytotoxic T cell markers and cytokines. Interesting preliminary data from the clinic have reported durable responses in two patients treated with pembrolizumab, harboring POLE mutations.71,72
Moreover, trials are underway to explore the use of immunotherapy based on CMS classification, aiming to overcome immunotherapy resistance mechanisms. Of note, the MoTriColor H2020 project is investigating atezolizumab combined with bevacizumab in patients defined as MSS by standard diagnostic tests, who present an “MSI-like” gene expression signature, classically within the CMS1 subtype. 73 The rationale comes from evidence of high TMB and dependence on angiogenic factors in in silico models. A positive outcome could amplify the percentage of CRC patients who may benefit from immunotherapy from 5% (MSI CRC) to 10%, opening the door to a more personalized use of immunotherapy strategies in molecularly selected patients (Table 6).
Other DNA repair deficiency alterations
Genomic and epigenomic events may be implicated in the DNA damage repair system in CRC. In vitro studies showed how CRC cells with ATM loss have defective homologous recombination and increased sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors. 74 Furthermore, O (6)-methylguanine-DNA-methyltransferase (MGMT) repair protein deficiency confers sensitivity to the cytotoxic alkylating drugs temozolomide and dacarbazine. Analysis of MGMT promoter hypermethylation by methyl-specific PCR was adopted in CRC due to the initial promising reports in glioblastoma. However, the results with alkylating agents in phase II clinical trials were poor. Recently, four phase II clinical trials with alkylating agents in chemotherapy-refractory mCRC tumors were conducted using a refined assessment of protein expression by IHC and percentage of promoter methylation by methyl-BEAMing (MP). The ORR exceeded 70%, with median PFS of 5 months. 75 The CAPTEM randomized phase II study aims to investigate the superiority of second-line capecitabine plus temozolomide compared with FOLFIRI in patients with RAS mutated, MGMT methylated tumors. No differences in survival and efficacy outcomes were observed for MGMT IHC-negative patients, indicating that phase III trials should explore alkylating agents in patients selected by IHC and MP. 76 Recently, the role of temozolomide on neoantigen load and induction of immune cell infiltration of the tumor microenvironment has been investigated in preclinical studies, showing that alkylating drugs promote a hyper-mutational status with increased renewal of neoantigens, and trigger long-lasting immune surveillance. An ongoing phase II study, ARETHUSA is exploring the use of temozolomide before an ICI in an MSS population. 77 Further lines of investigation are expected to confirm the rationale of chemotherapy plus immunotherapy combinations in molecularly defined mCRC populations.
Mesenchymal or “TGFβ active”
The CMS4 subgroup mesenchymal phenotype defines tumors with reduced sensitivity to chemotherapies and targeted strategies, such as the RAS WT CMS4 tumors intrinsically resistant to anti-EGFR drugs in preclinical studies and in retrospective analyses of clinical trials in which cetuximab is given in combination with oxaliplatin-based chemotherapies.78,79 Looking for actionable targets in this stromal-rich population is worthwhile given the high prevalence (close to 40%) of the mesenchymal signature in mCRC. 80
Preliminary in vivo studies are promising, with the inhibition of the TGFβ signaling preventing cross-talk between cancer cells and the microenvironment, and blocking tumor progression. 81 Furthermore, the use of chaperone (HSP90) inhibitors has been investigated with the aim of overcoming chemoresistance, and, more recently, of enhancing immunotherapy efficacy, based on the assumption of the strong immunosuppressive role of TGFβ in the tumor microenvironment of CMS4 tumors.82,83 TGFβ promotes T-cell exclusion and blocks the acquisition of the TH1-effector phenotype – a primary mechanism of immune evasion.
Tauriello et al. demonstrated how, after TGFβ inhibition, T cell infiltration increases in immunocompetent mice models, and that combining a TGFβ inhibitor with a PD-L1 blockade could overcome resistance to PD1 and lead to sustained antitumor immune responses. This key finding could be translated into the clinical setting for tumors considered to be immune “deserts”. 84
Implementation of the mesenchymal signature into clinical practice remains challenging due to the lack of affordable tests for paraffin tumor samples. Validation of new gene expression-based technologies in clinical molecularly selected trials is being investigated, such as in the MoTriColor project, where patients with mCRC testing positive for a “TGFβ active” gene expression signature in archived paraffin tissue are eligible to receive capecitabine in combination with the TGFβ inhibitor galunisertib.
The potential and current role of liquid biopsy in personalized treatment
In recent years, liquid biopsy allowed the integration into routine clinical care of assessing biomarkers of response with new technologies that track disease evolution in a reproducible manner. Hence, ctDNA represents a non-invasive tool to detect principal CRC driver mutations, and, therefore, provide a deeper understanding of the complex heterogeneity of CRC disease. Recent works have showed similar rate of detection of RAS mutation between plasma and tissue analyses, with a level of concordance close to 90%. 10 In addition, response to anti-EGFR mAbs could be efficaciously monitored in plasma, leading to the use of potential strategies as re-challenge treatments with cetuximab or panitumumab in patients with RAS WT status determined by ctDNA analysis (Table 2). However, researchers are facing the principal limits of liquid biopsy, including spatial and temporal heterogeneity that reduces concordance of CRC genotyping, even in the same individual, and the use of non-standardized techniques with different sensitivity, which determine conflicting results in clinical trials. For instance, as assessed before, the use of a determined cut-off to measure the RAS MAF in the blood of patients with mCRC is still lacking, oscillating between 1% and 5% resulting in a less than optimal patients’ selection. Undoubtedly, larger, prospective, multicenter clinical trials are urgently required to avoid discrepancies between techniques and cut-offs, with the final aim of ensuring the best approach is offered to every single patient with mCRC.
Conclusion
Our increasing genomic understanding of CRC complexity has led to major advances in the last few years, with the definition of the main drivers responsible for intrinsic and acquired resistance to standard and biological treatments. The ultimate goal is to facilitate larger clinical trials with biomarker-driven therapies that take into consideration the dynamic clonality of CRC and move the concept of “one marker – one drug” to a more integrated approach “multi-markers – drug combinations”.85,86 Undoubtedly, many issues need to be addressed. Transcriptomic analyses have enabled the identification of different molecular subtypes enriched with relevant biological characteristics, but the high rate of inter- and intratumor heterogeneity across various CRC subgroups limits the translation of the CMS classification in the clinic as a predictive marker.
The range of treatment options based on biomarker selection needs to be extended to a larger number of patients (e.g., immunotherapy strategies not only for the MSI population) and the analysis of the principal drivers of resistance to treatment should also be ensured at the time of the diagnosis of metastatic disease to identify oncogenic KRAS and NRAS before EGFR-targeted drugs, and BRAF mutations as early as possible in the disease history, as well as ERBB2 amplification in selected MSS RAS/BRAF WT tumors, left-sided, refractory patients who could participate in clinical trials. When feasible, comprehensive genomic tests should be performed to identify rare fusions, known to be enriched in RAS/BRAF WT MSI right-sided tumors. Likewise, NGS analysis for TMB quantification and DNA repair deficiency detection or gene expression profiling are reserved, in the current-day scenario, to selected research centers that could offer molecularly designed clinical trials, to identify oncogenic signatures of interest, such as “MSI-like” or mesenchymal phenotype. Making these techniques more widely available is important.
The future revolution in mCRC treatment will be fueled by a dynamic integration of genomic, transcriptomic, and immune-stromal characteristics to eliminate meaningless biomarkers and guarantee innovative effective therapeutic strategies.
Footnotes
Authors’ note
Giulia Martini and Davide Ciardiello are also affiliated with Università della Campania L. Vanvitelli, Naples.
Conflict of interest statement
GM, IB, JR, CD, JC, FS, NM declare no conflict of interest.
RD: advisory role for Roche; speaker’s fees from Roche, Symphogen, Ipsen, Amgen, Sanofi, MSD, Servier; and direct research funding from Merck.
GA: personal financial interests, honoraria for advisory roles, travel grants, research grants (past 5 years) from Hoffman La-Roche, Bristol-Myers Squibb, Bayer, Servier, Amgen, Merck Serono, Menarini; institutional financial interests, honoraria due to investigator contributions to clinical trials from Hoffman La-Roche, Novartis, Boehringer Ingelheim, Boston Pharmaceuticals, Hoffman La Roche, Genentech.
JT: personal financial interest in the form of scientific consultancy roles for Array Biopharma, AstraZeneca, Bayer, BeiGene, Boehringer Ingelheim, Chugai, Genentech, Inc., Genmab A/S, Halozyme, Imugene Limited, Inflection Biosciences Limited, Ipsen, Kura Oncology, Lilly, MSD, Menarini, Merck Serono, Merrimack, Merus, Molecular Partners, Novartis, Peptomyc, Pfizer, Pharmacyclics, ProteoDesign SL, Rafael Pharmaceuticals, F. Hoffmann-La Roche Ltd, Sanofi, SeaGen, Seattle Genetics, Servier, Symphogen, Taiho, VCN Biosciences, Biocartis, Foundation Medicine, HalioDX SAS and Roche Diagnostics; institutional financial interest in the form of financial support for clinical trials or contracted research for Agendia BV, Amgen SA, Debiopharm International SA, Janssen-Cilag SA, Mologen AG, Novartis Farmacéutica SA, Pharma Mar, Roche Farma SA, Laboratorios Servier SL and Symphogen A/S.
EE: personal financial interests, honoraria for advisory roles, travel grants, research grants (past 5 years) from Hoffman La-Roche, Sanofi Aventis, Amgen, Merck Serono, Servier, MSD, Array Pharmaceuticals, Bristol-Myers Squibb; institutional financial interests, honoraria due to investigator contribution in clinical trials from Hoffman La-Roche, Sanofi Aventis, Amgen, Merck Serono, MSD, Boehringer Ingelheim, AbbVie, Array Pharmaceuticals, Pierre-Fabre, Novartis, Bristol-Myers Squibb, GlaxoSmithKline, Medimmune.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.