
Abstract
Background:
There is an immediate need for research on the mechanism underlying telomerase activation and overexpression.
Materials & Methods:
A total of 174 patients with lung cancer (n = 106) and benign lung disease (n = 68) were recruited for the current study. The mRNA expression levels of E6, E7, LKB1, Sp1, and hTERC in brushing cells were detected by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR), and hTERC amplification was also detected by fluorescence in situ hybridization (FISH). To investigate the potential mechanism, bidirectional genetic manipulation was performed in well-established lung cancer cell lines.
Results:
Our results indicated that the mRNA expression levels of E6, E7, Sp1, and hTERC and the amplification level of hTERC were significantly increased in the malignant group compared with those of the benign group (p < 0.01). Conversely, the mRNA expression level of LKB1 was significantly decreased in the malignant group (p < 0.01). The correlation between E6, E7, Sp1, and hTERC expression was positive but was negative with LKB1 (p < 0.01). Our results also showed that HPV16 E6/E7 downregulated the expression of LKB1 at both the protein and mRNA levels. The loss of LKB1 upregulated Sp1 expression, and also promoted Sp1 activity. Sp1 further upregulated hTERC at the mRNA and gene amplification levels. Thus, we proposed a HPV–LKB1–Sp1–hTERC axis of E6/E7 upregulation of hTERC expression.
Conclusion:
We demonstrated for the first time that E6 and E7 promoted hTERC mRNA expression and the amplification of hTERC by relieving the effect of LKB1 on the phosphorylation of Sp1. Sp1 further activated hTERC by directly binding to the promoter regions of hTERC.
Keywords
Introduction
Telomerase is a ribonucleoprotein reverse transcriptase that contains an RNA subunit hTERC and a protein catalytic subunit hTERT. 1 In recent years, studies have found that the copy numbers of the TERT and TERC genes were substantially higher in most cancer cells than in normal cells. 2 Through their increased telomerase expression and activity, the vast majority of cancer cells acquire oncogenic changes to escape senescence, resulting in unlimited proliferation. Thus, understanding the mechanisms underlying telomerase activation and overexpression will be crucial for the identification of novel biomarkers, early disease detection, prognosis determination, and new therapeutic drug development. However, there are multiple mechanisms that are responsible for the upregulation of telomerase in cancer, and they are not completely understood. Molecular biology studies have found that high risk type HPV16 E6/E7 proteins are primary oncogenes, and a long-term persistent infection was capable of causing lung cancer.3–7 In our previous studies, we found high copy numbers of the hTERC gene due to its amplification in lung cancer brushing cells, 8 and we also found that E6/E7 inhibited the expression of the tumor suppressor gene LKB1,7–9 and that the loss of LKB1 upregulated Sp1 at both the protein and mRNA levels, as well as promoting Sp1activity. Sp1 further upregulated hTERT at both the protein and mRNA levels in two well-established lung cancer cell lines. 7 In the current study, we tried to investigate the relationship between HPV16 E6/E7 and the expression and amplification of the TERC gene, and the potential mechanisms that could be involved in this process were also explored.
Liver kinase B1 (LKB1) is a tumor suppressor gene and regulates gene expression by phosphorylating substrate proteins or binding to target proteins. LKB1 plays an important role in the development of lung cancer.10–12 Liang et al. reported that the overexpression of the LKB1 protein downregulated the expression and activity of transcription factor specificity protein 1 (Sp1). 12 We also found that LKB1 had a significant inhibitory effect on the expression of Sp1 at the protein level, mRNA level, and transcriptional activity level. 7
Sp1, a member of the Sp proteins family, constitutes a group of highly conserved transcription factors that are present in a wide range of organisms. Their structures are defined by the presence of three highly conserved DNA-binding zinc finger domains that bind to similar, yet distinct, GC-rich target sequences. Recently, we found that Sp1 mRNA is overexpressed in the bronchial brushing cells of patients with lung cancer. 13 Hedrick et al. demonstrated that the selective knockdown of Sp1 by RNAi in the A549 lung cancer cell line resulted in the inhibition of cell growth, decreased survival, and inhibition of migration/invasion. 14 We also found that the specific knockdown of Sp1 by RNAi in lung cancer cells had a significant inhibitory effect on the expression of hTERT at the protein level and mRNA level. 7
In the current study, we aimed to explore the role played by E6, E7, LKB1 and Sp1 in the regulation of hTERC expression in lung cancer cells. We demonstrated that HPV16 E6/E7 inhibited the expression of LKB1 and that the loss of LKB1 upregulated the expression of Sp1; Sp1 further upregulated hTERC at the mRNA and gene amplification levels. More interestingly, we found that LKB1 inhibited Sp1 activity by promoting Sp1 phosphorylation. Furthermore, Sp1 upregulated the mRNA expression of hTERC by activating the hTERC promoter regions.
Materials and methods
The study was conducted according to the guidelines of the institutional review boards at the First Affiliated Hospital of China Medical University. We obtained the internal review board approval (no. 2013-17, China Medical University) and informed consent of patients for this study. The brushing cells from 106 lung cancer patients who attended the laboratory of cytopathology at the First Hospital of China Medical University during the period of 2013–2014 were randomly collected in the study. There were 88 men (83.0%) and 18 women (17.0%) in the study, with a mean age of 64.3 years (range 40–79). Of the malignant cells, 20 were adenocarcinomas (ACs), and 86 were squamous cell carcinomas (SCCs). The unbalance between adenocarcinoma and SCC subtypes in the study population is due to the fact that bronchoscopies are possible for patients with central lung cancer, and SCC is the most common histological type of central lung cancer, only a small percentage of adenocarcinomas are central lung cancers. Another group of 68 randomly selected patients without lung cancer were included as controls (58 with inflammation and 10 with endobronchial tuberculosis). All 68 of the control patients had biopsies, resections or clinical follow-up results that were negative for malignancy. All bronchoscopies were performed by two experienced bronchoscopists. Detailed procedures for bronchoscopy and the cytological diagnosis are described in the reference with PMID 28813465. 7 The specimen we applied for RNA extraction and fluorescence in situ hybridization (FISH) test in this study were rest abandoned specimens after pathological examination, no medical operations beyond routine medical examination has been undertaken in the process of drawing material, the rest of the specimen was of no other use even if not used in this study.
Cell culture
Based on our previous screening results in lung cancer cell lines, H1299 and H460 were selected as being representative of E6 or E7-low cell lines, respectively, whereas A549 and LK2 were selected as representative of E6 or E7-high cell lines, respectively. These cell lines were selected for the following transfection and interference assays. The A549, H1299 and H460 cell lines were obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA). The LK2 cell lines were obtained from the Cell Bank of the Chinese Academy (Shanghai, China). The A549 and LK2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM); other cells were cultured in RPMI-1640 that was supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% carbon dioxide (CO2) humidified atmosphere.
Plasmid construction and transfection
HPV16 cDNA (p-EGFP-N1-HPV16E6/E6mut, p-EGFP-N1-HPV16E7/E7mut and p-EGFP-N1, gifts from Prof. Xudong Tang, Institute of Biochemistry and Molecular Biology, Guangdong Medical College, China) was transfected into H1299 and H460 cell lines which expresses a relatively low level of HPV16. 6 LKB1 (pcDNA3-LKB1-Hisand pcDNA3-His, gifts from Prof. Xin Hou, College of Life Sciences, Inner Mongolia University, Huhhot, Inner Mongolia, China) was transfected into A549 and LK2 cell lines, which expresses a relatively low level of LKB1. 15 The mutants and empty plasmids were used as negative controls. Cells that were exposed to Lipofectamine 2000 or Oligofectamine alone served as mock transfection controls. The transfection efficiency was evaluated by observing green fluorescence under a fluorescence microscope and with a flow cytometric analysis (Epics-XL, Coulter, USA).
Small interfering RNA
Short-interference RNAs (siRNAs) against E6, E7, LKB1, and Sp1 were purchased from RIBOBIO (Guangzhou, China). Scrambled siRNA (RIBOBIO, Guangzhou, China) was used as a non-specific siRNA control. For the siRNA transfection, cells were seeded at 5 × 104 cells/35-mm well. Then, 24 hours later, the siRNA was transfected into the cells using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA). After transfection, the cells were incubated for 48 hours and subjected to various analyses.
Western blot analysis
The assays were performed as previously described. 6 Information about primary antibodies is as follows: HPV16 E6 (1:200, Bioss Biotechnology Co., Ltd., Beijing, China), HPV16 E7 (1:200, Bioss Biotechnology Co., Ltd., Beijing, China), LKB1 (1:1000, Cell Signaling Technology, Danvers, MA, USA), Sp1 (1:1000, Cell Signaling Technology, Danvers, MA, USA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000, Cell Signaling Technology, Danvers, MA, USA).
Real-time PCR
The assays were performed as previously described. 7 Detailed information on the primers is provided in Table 1.
Sequences and features of primers used for qRT-PCR.
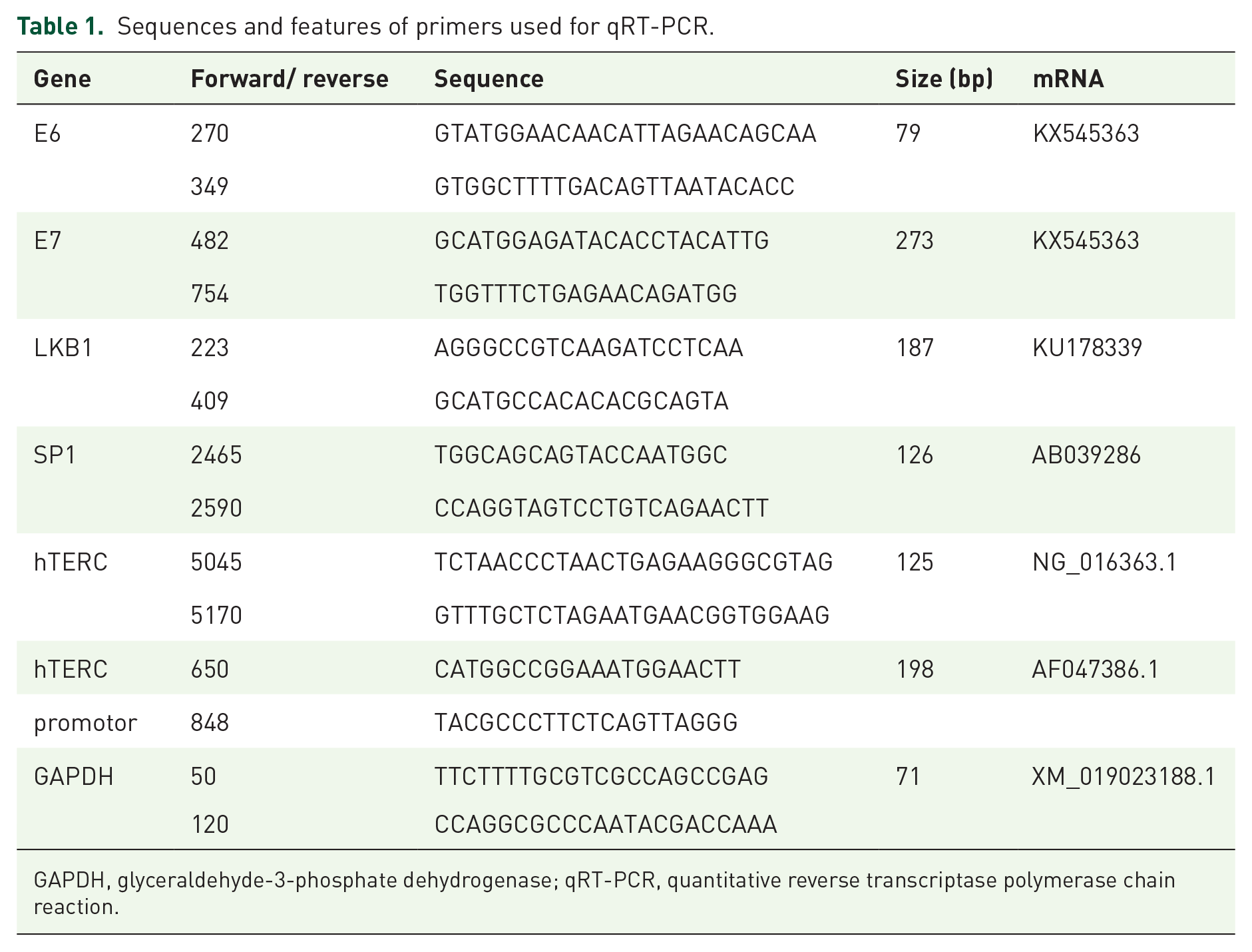
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; qRT-PCR, quantitative reverse transcriptase polymerase chain reaction.
Luciferase reporter assay
The H1299 cell line and A549 cell line were seeded at 70–80% confluence in a 24-well plate one day before they were co-transfected with the firefly luciferase reporter (0.2 µg) along with the Renilla luciferase reporter (Promega, Madison, WI, USA) (0.02 µg) using Lipofectamine 3000 (Invitrogen, USA) for 6 hours in the transfection medium. After replacing the transfection medium with complete medium, the cells were incubated for 24 hours. Cell lysates were analyzed for their luciferase activities using the Dual-Luciferase Assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The pGMSP1-Lu was purchased from Genomeditech, China. Relative luciferase activity was calculated by normalizing the ratio of Firefly/Renilla luciferase activity to that of the negative control-transfected cells.
Chromatin immunoprecipitation assay (ChIP)
An EZ-ChIP Chromatin Immunoprecipitation Kit (Millipore, MA, USA) was used according to the instructions that were recommended by the manufacturer. Briefly, lung cancer cells were cultured at a density of 106–107 in 100-mm dishes, the chromatin was cross-linked with fresh formaldehyde, and glycine was used for removing the cross-linking. Sonication was used to break the cross-linked chromatin, and the average length of the fragments was between 200 bp and 600 bp. Then, chromatin immunoprecipitation was performed with 10 µg Sp1 antibody or the negative control IgG antibody that was included in the kit. From each group, 1/10 of the lysate volume was removed from the samples to detect the total chromatin content. The precipitated DNA was purified and used as a template for PCR to amplify the promoter region of hTERC. Primer3 was used to design the primers for the hTERC promoter PCR, and the primers that were used are listed in Table 1.
Fluorescence in situ hybridization
The cells were coated, air-dried, then fixed with ethanol; after this, the cells were digested by pepsin (0.1 g pepsin powder per 40 mL 0.01 M hydrochloric acid (HCL)) for 7 minutes at 37°C, and then the cells were washed twice in 2 × saline sodium citrate (SSC). 4′,6-diamidino-2-phenylindole (DAPI) was added to the slides to observe the nuclear content. After they were soaked in 2 × SSC for 5 minutes, the slides were dehydrated and air-dried. Then, 10 µL of probe was added and denatured for 5 minutes at 75°C, after which the hybridization was performed at 42°C overnight. The post-hybridization wash was performed three times in 2 × SSC at 56°C for 5 minutes. The slides were then fixed in 70% ethanol. After air-drying in the dark, the slides were cover slipped and observed using an Olympus fluorescent microscope (BX-51; Olympus, Tokyo, Japan) that was equipped with a X-cite 120 mercury lamp. The probe panel (F01007-00; China Medical Technologies, Beijing, China) that was used consisted of two probes: hTERC (red) and the control, centromeric chromosome 3 (green). We counted the signal as previously described. 8
Statistical analyses
The SPSS 16.0 statistical software package (SPSS, Inc., Chicago, IL, USA) was used for all analyses. A Student’s t-test was used to compare the data from the densitometry analysis. McNemar’s test was used to compare the mRNA expression of E6, E7, LKB1, Sp1, and hTERC in benign and malignant brushing cells. A Spearman correlation analysis was used to determine the correlation among the mRNA expression of E6, E7, LKB1, Sp1, and hTERC in the malignant group. All data were presented as the means ± SD of in vitro experiments that were performed at least three times. The level of statistical significance was set at p < 0.05.
Results
The quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) and FISH analyses of the brushing cells of patients with benign or malignant lung diseases, including AC and SCC, are presented in Table 2.
The mRNA of E6, E7, LKB1, Sp1 and hTERT with qRT-PCR as well as the amplification of hTERC with FISH in bronchial brushing cells of patients with benign and malignant lung lesions (mean ± SEM).

AC, adenocarcinoma; E6, human papillomaviruses 16 E6; E7, human papillomaviruses 16 E7; FISH, fluorescence in situ hybridization; hTERC, RNA component of human telomerase gene; LKB1, Liver kinase B1; qRT-PCR, quantitative real time reverse transcriptase polymerase chain reaction; SCC, squamous cell carcinoma; Sp1, specificity protein 1.
p < 0.01 as compared with benign.
The mRNA expression levels of E6, E7, Sp1, and hTERC and the amplification level of hTERC were significantly increased in the malignant group compared with the benign group (p < 0.01). Conversely, the mRNA expression level of LKB1 was significantly decreased in the malignant group (p < 0.01). The correlation analyses between E6, E7, Sp1, hTERC, and LKB1 were performed, and our results indicated that the correlations between E6, E7, Sp1, and hTERC were positive, but there was a negative correlation with LKB1 (p < 0.01).
The overexpression of both E6 and E7 significantly downregulated the expression of LKB1, but upregulated the expression of Sp1 and hTERC, as well as the transcriptional activity of Sp1 in the H1299 and H460 cells; these results are presented in Figure 1a and b as well as Figure 2a and b.

The overexpression of E6, E7, Sp1, and hTERC but low expression of LKB1 was observed in H1299 (a) and H460 (b) cells, whereas low expression of E6, E7, Sp1, and hTERC but overexpression of LKB1 was observed in A549 (c) and LK2 (d) cells. Detection of both protein and mRNA expression of E6, E7, LKB1, and Sp1 by western blot and qRT-PCR, of the mRNA expression of hTERC by qRT-PCR, and of the transcriptional activity of Sp1 by a luciferase reporter was performed in lung cancer cells as well as in control cells.
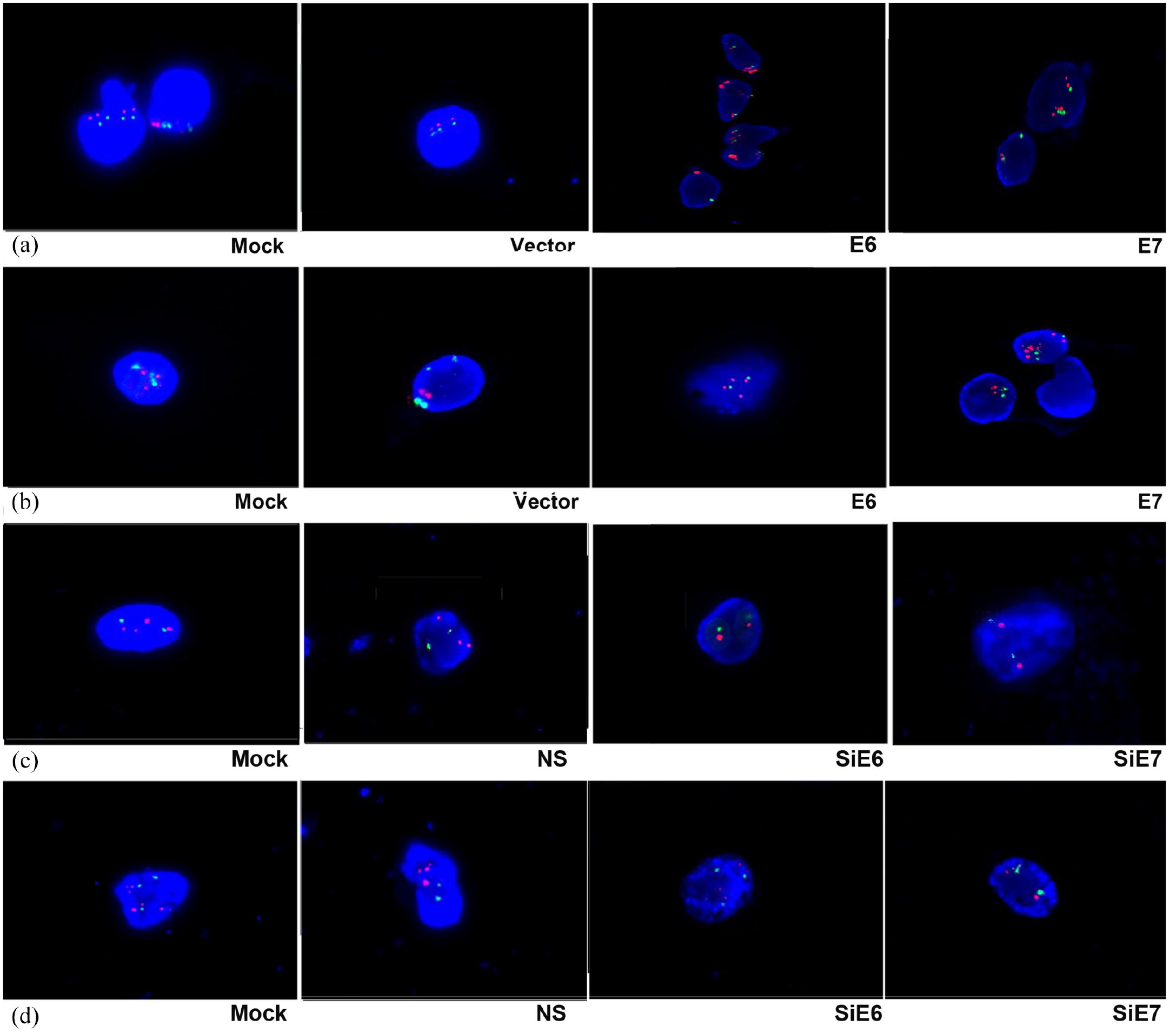
The overexpression of E6 and E7 was observed in H1299 (a) and H460 (b) cells, and the interphase FISH analysis shows abnormal signal patterns with amplification of hTERC compared with Mock or Vector (×1000). The low expression of E6 and E7 was observed in A549 (c) and LK2 (d) cells, and the interphase FISH analysis shows normal signal patterns without amplification of hTERC compared with Mock or NS (×1000).
The pEGFP-N1-E6 or E7 vectors were transiently transfected into the two low expression H1299 and H460 cell lines, and the E6 or E7 empty vectors and mock transfections served as controls. The results showed that the overexpression of E6 or E7 significantly downregulated the expression of LKB1 at the protein level and at the mRNA level but upregulated the expression of Sp1 at the protein level and at the mRNA level; as well as the expression of hTERC at the mRNA level and at the amplification level. The transcriptional activity of Sp1 was also upregulated.
The inhibition of both E6 and E7 upregulated the expression of LKB1 but downregulated the expression of Sp1 and hTERC as well as the transcriptional activity of Sp1 in the A549 and LK2 cells; these results are presented in Figure 1c and d as well as Figure 2c and d.
To verify further the regulatory roles of both E6 and E7 on LKB1, Sp1, and hTERC, we applied E6 or E7-specific siRNA to knock down the expression of E6 or E7 in the A549 and LK2 cell lines, and we used E6 or E7-non-specific siRNA and mock specific siRNA to serve as the controls. The results indicated that the inhibition of both E6 and E7 upregulated the expression of LKB1 at the protein and mRNA levels, but downregulated the expression of Sp1at the protein level and at the mRNA level, as well as the expression of hTERC at the mRNA level and at the amplification level. The transcriptional activity of Sp1 was also downregulated. E6 or E7-non-specific siRNA and mock specific siRNA showed minimal or no change.
LKB1 negatively regulates the expression of Sp1 and hTERC as well as the transcriptional activity of Sp1, and it also promotes Sp1 phosphorylation.
To elucidate the regulatory mechanism of E6 and E7 on hTERC and to explore the regulatory role of LKB1 on Sp1 and hTERC in lung cancer cells, we selected H1299 and H460 cells, which have high LKB1 expression, and A549 and LK2 cells, which have low LKB1 expression. The results showed that the overexpression of LKB1significantly downregulated the expression of Sp1 at the protein level and at the mRNA level, and it also downregulated the expression of hTERC at the mRNA level. The transcriptional activity of Sp1 was also downregulated (Figure 3a–b). Conversely, the results of LKB1 inhibition are shown in Figure 3c–d.

The overexpression of LKB1 but low expression of Sp1 and hTERC was observed in A549 (a) and LK2 (b) cells, whereas the low expression of LKB1 but overexpression of Sp1 and hTERC was observed in H1299 (c) and H460 (d) cells. Detection of both protein and mRNA expression of LKB1 and Sp1 by western blot and qRT-PCR, of the mRNA expression of hTERC by qRT-PCR, and of the transcriptional activity of Sp1 by a luciferase reporter assay was performed in lung cancer cells as well as in control cells.
HPV16 E6/E7 relieved the effect of LKB1 on Sp1 phosphorylation
We transfected E6 or E7 plasmids into the H1299 and H460 cell lines, and then collected the cells at five consecutive time points (0, 12, 24, 36, and 48 hours). Western blots were performed for E6/E7, LKB1, pSp1 (T739) and pSp1 (T453). The results showed that as the expression levels of E6 or E7 increased, the expression levels of LKB1 decreased, and the phosphorylation levels of Sp1 at Thr739 and Thr453 were also decreased (Figure 4a–d).
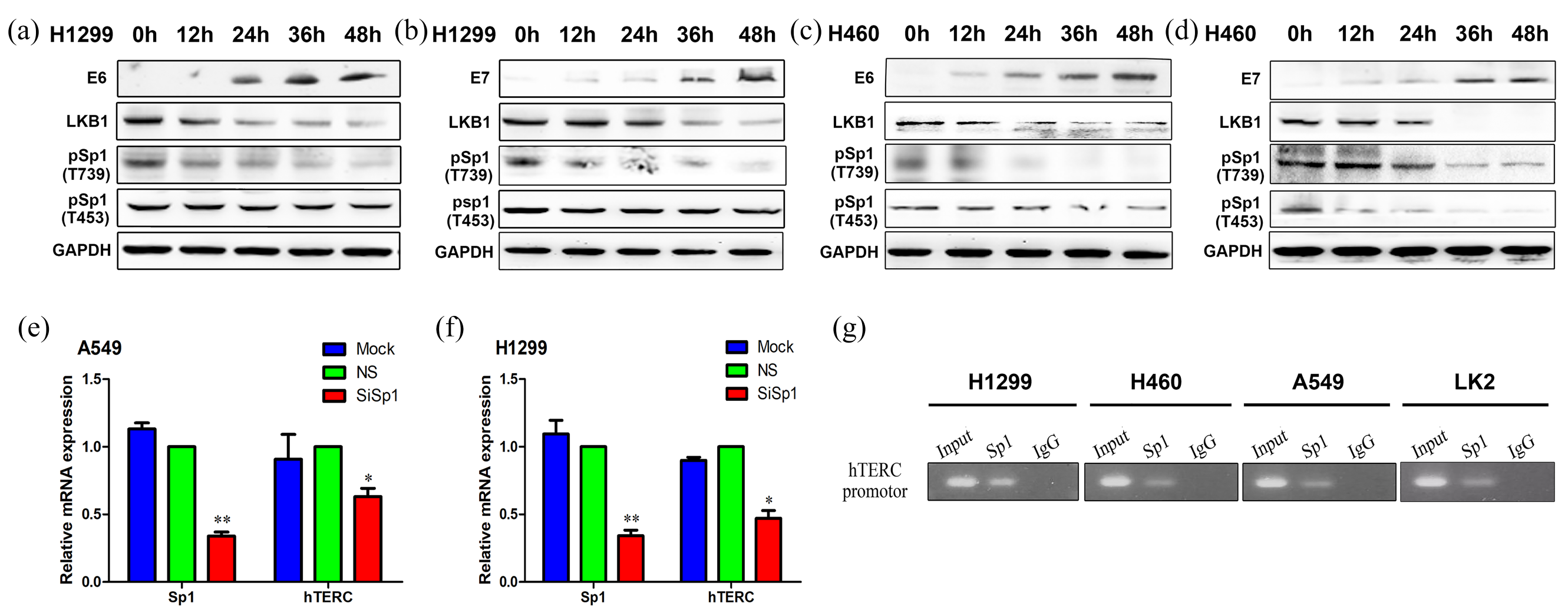
The overexpression of E6 or E7 but low expression of LKB1, pSp1 (T739), and pSp1 (T453) were measured at 0, 12, 24, 36, and 48 hours in H1299 (a or b) and H460 (c or d) cells, and the protein expressions of E6, E7, LKB1, Sp1, pSp1 (T739), and pSp1 (T453) were detected by western blot in lung cancer cells. The low expression of Sp1 and hTERC was observed in A549 (e) and H1299 (f) cells, and the mRNA expression levels of both Sp1 and hTERC were quantified by qRT-PCR in lung cancer cells. The binding between Sp1 and the hTERC promoter region was shown by chromatin immunoprecipitation in H1299, H460, A549, and LK2 cells (g).
Sp1 upregulated the mRNA expression of hTERC by activating the hTERC promoter regions
Interfering with the expression of Sp1 in the H1299 and A549 cells resulted in the significantly decreased mRNA expression of hTERC (Figure 4e–f). With the application of JASPAR software, we found a possible direct binding site of Sp1 to the promoter of hTERC with a sequence of GTCCTTCCTCA (Gene ID: AF047386.1; hTERC promoter: 1–791; binding site: 641–651). The sequence was further verified by chromatin immunoprecipitation. The result is shown in Figure 4g.
Discussion
Bronchial brushing is an important method for the cytodiagnosis of lung cancer, and it is also an important supplement to the histodiagnosis of a forceps biopsy.15–17 Recently, we demonstrated that qRT-PCR from bronchial brushing specimens increased the sensitivity of diagnoses.13,18–20 In current study, we evaluated the relationship between HPV16 E6/E7 and the hTERC gene in a total of 174 patients that included patients with lung cancer (n = 106) and benign lung disease (n = 68). Of the patients with lung cancer, 20 were ACs, and 86 were SCCs. The unbalance between adenocarcinoma and SCC subtypes in the study population is due to the fact that bronchoscopies are possible for patients with central lung cancer, and SCC is the most common histological type of central lung cancer, only a small percentage of adenocarcinomas are central lung cancers. The mRNA expression levels of E6, E7, LKB1, Sp1, and hTERC in the brushing cells were detected by qRT-PCR, and the hTERC amplification level was detected by FISH. Our results showed that the mRNA expression levels of E6, E7, Sp1, and hTERC and the amplification level of hTERC were significantly increased in the malignant group, compared with the benign group (p < 0.01). Conversely, the mRNA expression level of LKB1 was significantly decreased in the malignant group (p < 0.01). The correlations between E6, E7, Sp1, hTERC, and LKB1 were analyzed, and our results indicated that the correlations between E6, E7, Sp1, and hTERC were positive but were negative with LKB1 (p < 0.01). These results indicate that there are regulations among E6, E7, LKB1, Sp1 and hTERC.
The potential molecular mechanisms that were involved in this process were further explored. Recent studies by us and other investigators have shown that chronic infection by HPV16 E6/E7 inhibited the expression of LKB1 in lung cancer specimens.7,9,21 In this study, we confirmed that the overexpression of E6 and E7 in lung cancer cells downregulated the expression of LKB1 at both the protein and mRNA levels. Interestingly, the knock down of LKB1 upregulated Sp1 expression at both the protein and mRNA levels, and it also promoted Sp1 activity by decreasing Sp1 phosphorylation at Thr739 and Thr453. Sp1 subsequently upregulated hTERC expression at the mRNA level and gene amplification level. These results demonstrated that Sp1 and hTERC are downstream effectors of LKB1. Thus, E6/E7 upregulated the expression of hTERC through the HPV–LKB1–Sp1–hTERC axis (Supplementary Figure 1). Sp1 regulates activation of many genes implicated in tumor growth and cell cycle progression. The phosphorylation of Sp1 at Thr739 inhibited the DNA binding ability and the transcriptional activity of Sp1. 22 The simulated phosphorylation at Thr739 and Thr453 inhibits half of the transcriptional activity of Sp1. 23 In this study, we identified a novel substrate of LKB1: Sp1. We hold the opinion that the inhibition of the transcriptional activity of Sp1 by LKB1 partly depends on the phosphorylation at Thr739 and Thr453. However, further studies are still required to confirm the existence of other vital sites of Sp1, which could be phosphorylated by LKB1.
HPV16 E6/E7 was also noted to relieve the effect of LKB1 on Sp1 phosphorylation. We transfected E6 or E7 plasmids into H1299 and H460 cell lines and then collected the cells at five consecutive time points (0, 12, 24, 36, and 48 hours). Western blot results for E6/E7, LKB1, pSp1 (T739) and pSp1 (T453) showed that, as the expression levels of E6 or E7 increased, the corresponding expression levels of LKB1 decreased and the phosphorylation levels of Sp1 at Thr739 and Thr453 were decreased as well.
Zhao JQ et al. showed that multiple signals including Sp1, Sp3, pRB, and NF-Y contributed to the regulation of hTERC gene expression. 24 These factors activated or suppressed the activity of hTERC by directly binding to the hTERC promoter regions. With JASPAR software, we found the possible direct binding site of Sp1 to the promoter of hTERC, which had a sequence of GTCCTTCCTCA (Gene ID: AF047386.1; hTERC promoter: 1–791; binding site: 641–651). The sequence was further verified by chromatin immunoprecipitation. Our results indicated that Sp1 upregulated the mRNA expression of hTERC by activating the hTERC promoter regions.
In conclusion, we demonstrated that the expression of E6, E7, Sp1, and hTERC mRNAs were increased in brushing cells of lung cancer patients when compared with those obtained from patients with benign diseases (p < 0.01). The correlation comparisons between E6, E7, Sp1, and hTERC were positive but were negative with LKB1 (p < 0.01). For the first time, we demonstrated that E6 and E7 promoted hTERC mRNA expression and the amplification of hTERC by relieving the effect of LKB1 on the phosphorylation of Sp1. The overexpression of LKB1 significantly upregulated the phosphorylation levels of Sp1 at Thr739 and Thr453. Sp1 further activated hTERC by directly binding to the promoter regions of hTERC. These results indicated that both E6 and E7 play a predominant role in regulating the gene expression of hTERC. Thus, understanding of the molecular basis of hTERC gene regulation may be crucial for the identification of novel biomarkers, early disease detection, and new therapeutic drug development for HPV-related cancer.
Supplemental Material
S1_Fig – Supplemental material for HPV16 E6/E7 upregulate hTERC mRNA and gene amplification levels by relieving the effect of LKB1 on Sp1 phosphorylation in lung cancer cells
Supplemental material, S1_Fig for HPV16 E6/E7 upregulate hTERC mRNA and gene amplification levels by relieving the effect of LKB1 on Sp1 phosphorylation in lung cancer cells by Jing-Hua Yang, Ming-Zhe Wu, Xu-Bo Wang, Shiyu Wang, Xue-Shan Qiu, En-Hua Wang and Guang-Ping Wu in Therapeutic Advances in Medical Oncology
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China to Guang-Ping Wu, grants nos. 81171650 and 81672082.
Supplementary material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.