
Abstract
Background:
The objectives of this phase II study were to determine the clinical activity of the MET tyrosine kinase inhibitor capmatinib (INC280) in patients with MET-dysregulated advanced hepatocellular carcinoma (HCC) and to assess the safety, pharmacokinetics, and correlation of biomarkers with the response.
Methods:
This phase II, open-label, single-arm study evaluated twice daily (BID) oral capmatinib in a dose-determining stage, utilizing a Bayesian Logistic Regression Model (BLRM) subject to Escalation with Overdose Control criteria, safety, pharmacokinetics, and pharmacodynamic information to determine a recommended dose for expansion (RDE) evaluating efficacy in patients with MET-dysregulated HCC.
Results:
A total of 38 patients received treatment. In the dose-determining stage, patients received capmatinib 300 mg BID capsules (n = 8), and in the expansion, patients received 600 mg BID capsules (n = 28) or 400 mg BID tablets (n = 2) based on the BLRM and other relevant clinical data. No predefined qualifying adverse events (AEs) were observed during the first 28 days of treatment, and the RDE was 600 mg BID capsules (equivalent pharmacokinetics to 400 mg BID tablets). The most common any causality AEs were nausea (42%), vomiting (37%), and diarrhea (34%). In the expansion stage, in a subgroup of 10 patients with MET-high HCC, the overall response rate was 30%, including 1 durable complete response (>600 days) and 2 partial responses [1 durable (>600 days)].
Conclusions:
Single agent capmatinib at the RDE is tolerable with a manageable safety profile. Antitumor activity was seen in a subset of patients with MET-dysregulated (MET-high) HCC.
Trial registration:
ClinicalTrials.gov: NCT01737827. https://clinicaltrials.gov/ct2/show/NCT01737827
Introduction
According to the World Health Organization, hepatocellular carcinoma (HCC) is the fifth most common malignancy and the second major cause of tumor-related death in the world today. 1 Although HCC is being diagnosed earlier, patients with advanced HCC have poor long-term survival, and the incidence and mortality rates are rising.2,3 Activation (overexpression, amplification, or both) of the MET signaling pathway [where the MET gene encodes MET/hepatocyte growth factor (HGF) receptor protein] has been observed in 20–48% of patients with HCC, 4 which was determined using a variety of methods, including greater than median density by Western blotting, increased MET gene expression signature, MET copy number (CN) gain and mRNA expression, and positive (>20% of tumor section) immunohistochemistry (IHC) staining.4–9 In addition, overexpression by these criteria was shown to predict shorter survival in patients with HCC.4–7 The MET receptor tyrosine kinase binds its sole ligand HGF, which then activates the RAS mitogen-activated protein kinase (MAPK) pathway, phosphatidylinositol-3 kinase (PI3K)-protein kinase B (PKB or AKT) pathway, mammalian target of rapamycin pathway, signal transducer and activator of transcription (STAT) pathway, beta-catenin pathway, and Notch pathway. Activation of the MET signaling pathway, therefore, promotes cell proliferation, survival, and metastasis.10,11 Experimental evidence demonstrated that MET inhibition abrogates the growth of MET-activated HCC cells by blocking MET phosphorylation and the activation of the downstream PI3K and MAPK pathways. 12 In addition, overexpression of HGF and MET amplification has been shown to predict the sensitivity of human HCC xenografts to MET inhibition. 13
Capmatinib (INC280) is a highly potent and selective MET inhibitor in biochemical and cellular assays and causes regression of MET-dependent tumor models in animals at well tolerated doses. 14 In addition, MET-amplified experimental HCC tumors have been shown to be highly sensitive to capmatinib. 13 In a phase I clinical study carried out in patients with advanced solid tumors, a recommended phase II dose (RP2D) of 600 mg twice daily (BID; capsule) and 400 mg BID (tablet) was identified, and capmatinib was shown to have a tolerable safety profile.15–17 Preliminary antitumor efficacy was reported in patients with MET-dysregulated non-small cell lung cancer, in particular, patients with a high level of MET amplification, MET exon 14 deletion mutation, or both.15,16 This study is a phase II, open-label, single-arm, multicenter study of capmatinib in patients with advanced HCC and confirmed MET pathway dysregulation who have received no prior systemic therapy [ClinicalTrials.gov identifier: NCT01737827].
Patients and methods
Study oversight
This study was performed in accordance with the Declaration of Helsinki and the principles of Good Clinical Practice. The protocol was approved by an Institutional Review Board at each investigative site (Supplementary Table 1), and all patients provided written informed consent before any study procedures. The study was designed by the sponsor (Novartis Pharmaceuticals Corporation). The sponsor collected the data and analyzed them in conjunction with the authors.
Study design
This phase II dose-determining and expansion study planned to enroll approximately 56 patients from the Asia-Pacific region with advanced HCC. The primary objective was to determine the overall clinical activity of capmatinib in patients with advanced HCC and confirmed MET dysregulation, with a primary endpoint of time to progression (TTP). However, due to the difficulty in identifying eligible patients, the study enrollment was halted prior to the completion of the expansion stage. Kaplan–Meier analysis of this endpoint was not performed due to the insufficient sample size for a meaningful estimate. Secondary objectives reported were the further assessment of the clinical activity of capmatinib in patients with advanced HCC and MET dysregulation, with secondary endpoints of overall response rate and disease control rate. Other secondary objectives included the evaluation of safety, pharmacokinetics (PK), and assessment of correlation of serum HGF levels with clinical response. Eligible patients were aged ⩾18 years old with advanced HCC not suitable for, or which progressed following locoregional therapy. In addition, patients were required to have a current cirrhotic status of Child–Pugh class A with no encephalopathy, and Eastern Cooperative Oncology Group (ECOG) performance status of 0−2. Key exclusion criteria included prior systemic chemotherapy or molecular-targeted therapy for HCC, previous treatment with MET-targeted or HGF-targeted therapy, previous local therapy completed <4 weeks prior to dosing and, if present, any related acute toxicity greater than grade 1, known active bleeding within 2 months prior to screening or with history or evidence of inherited bleeding diathesis or coagulopathy, or clinically significant venous or arterial thrombotic disease within the past 6 months.
It is important to highlight that the definition of MET positivity evolved throughout the duration of this study. MET positivity was originally defined as MET H-score ⩾50 or ratio of MET gene copy number/centromeres ⩾2.0 or MET (GCN) ⩾5. This less stringent definition of MET positivity was based upon an observation of a near-universal upregulation of the MET ligand HGF in tumor-adjacent liver tissue obtained from patients with HCC, suggesting a potential mechanism for MET activation even at medium expression levels in HCC tumors. Based on preliminary data from capmatinib clinical studies that indicate that high-MET protein expression and increased MET GCN may be predictive of response to capmatinib,15–18 a protocol amendment with revised biomarker inclusion criteria was implemented after 33 patients had been enrolled, including 20 patients in the dose-expansion stage of the study who did not meet the new criteria. The original eligibility criteria were revised to specify that tumors must have MET-high status defined as a MET IHC intensity score of 3+ in ⩾50% tumor cells or 2+ in ⩾50% of tumor cells plus MET GCN ⩾5 by fluorescence in situ hybridization (FISH). Patients with tumors showing MET GCN ⩾5 by FISH, but with IHC data unavailable for technical reasons, were also eligible. Patients who met these new MET positivity criteria are referred to as ‘MET-high’ in the manuscript to differentiate from the previous definition.
Treatment plan and drug administration
Capmatinib was administered orally at a starting dose of 300 mg BID (capsules) in the dose-determining stage of the study, a Bayesian Logistic Regression Model (BLRM), in combination with available safety information and pharmacodynamic (PD), PK information, or both, were utilized to determine a recommended dose for the expansion phase. In the expansion phase, patients were treated with 600 mg BID capsules or 400 mg BID tablets, which are pharmacokinetically equivalent. Patients were treated with capmatinib by dosing continuously in 21-day cycles. Treatment was continued until either disease progression [per Response Evaluation Criteria in Solid Tumors (RECIST 1.1)] as determined by the investigator, unacceptable toxicity that precluded further treatment, pregnancy, discontinuation at the discretion of the investigator or patient, withdrawal of consent, loss to follow-up, or death.
Assessments
Antitumor activity
Clinical efficacy assessments were based on radiographic tumor measurements (RECIST 1.1). Computed tomography-based tumor assessments were performed unless contraindicated, in which case MRI with contrast was performed.
Safety
Safety assessments were carried out based on adverse events (AEs) graded according to the National Cancer Institute Common Terminology for Adverse Events and physical examination, electrocardiogram, performance status, and laboratory evaluations.
Pharmacokinetics
PK assessments during the dose-determining stage were based on full PK blood samples collected from days 1–2 and days 15–16 in cycle 1, and predose blood samples were collected on cycle 2 day 1 and cycle 3 day 1. Plasma PK parameters were determined using noncompartmental methods. During dose-expansion, limited PK blood samples were collected predose and postdose on cycle 1 day 1, cycle 1 day 15, cycle 2 day 1, and cycle 3 day 1. Capmatinib concentrations were measured in plasma using liquid chromatography-tandem mass spectrometry.
Exploratory biomarker analysis
Serum HGF levels were quantified at variable time points using enzyme-linked-immuno-sorbent assay. Next-generation sequencing (Foundation Medicine Inc., Cambridge, MA, USA) was carried out (T5a test on 287 genes and 19 gene rearrangements) on available screening and cycle 1 day 15 tumor samples.
Results
Patient demographics and disposition
Between 25 March 2013 and the primary analysis cut-off date of 28 February 2017, a total of 38 patients were treated. In the dose-determining stage, patients were treated with capmatinib 300 mg BID capsules (n = 8), and in the dose-expansion stage, patients were treated with 600 mg BID capsules (n = 28) or 400 mg BID tablets (n = 2) based on the BLRM and other relevant clinical data, including the RP2D that was determined in a phase I study in patients with advanced solid tumors.15–17 All patients were Asian, the majority with moderately to poorly differentiated HCC, with a mean age of 55.6 years, 89% were men, 53% had an ECOG performance status of 0, and distant metastases were present in 39% of patients (Table 1). Overall, 12/38 patients (2 patients in the 300 mg BID capsule dose-escalation group, 8 patients in the 600 mg BID capsule expansion group, and 2 patients in the 400 mg BID tablet expansion group) had tumors characterized as MET-high (MET IHC intensity score 3+ in ⩾50% tumor cells, or 2+ in ⩾50% of tumor cells plus MET GCN ⩾5 by FISH). At the time of data cut-off, 36/38 patients discontinued treatment (61% due to disease progression; 24% due to AEs), and 2 patients were receiving ongoing treatment.
Baseline demographics and prognostic factors for HCC (full analysis set).

AFP, alpha fetoprotein; BID, twice daily; ECOG PS, Eastern Cooperative Oncology Group performance status; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; SD, standard deviation.
Treatment exposure
The median duration of exposure to capmatinib was 55.5 days (10−718 days), with 79% of patients treated for >3 weeks, and 18% receiving treatment for > 18 weeks. The duration of exposure and overall response (RECIST 1.1; investigator assessed) are presented in Figure 1. Dose reduction (not including formulation change) was required in 11/38 (29%) patients, 7/38 (18%) patients had at least 1 dose reduction due to AEs.

Duration of exposure and overall response (per RECIST v1.1; investigator assessed), dose-expansion stage (full analysis set).
Dose and formulation determination
In the dose-determining stage of the study, eight patients were enrolled and treated with 300 mg BID capsules and six patients were evaluable for dose decision analysis guided by the BLRM [subject to escalation with overdose control (EWOC)criteria], PK/PD, clinical, and laboratory factors. No qualifying AEs (predefined AEs or abnormal laboratory values assessed by the investigator as related to therapy with capmatinib) were observed in any of these patients, and based on the BLRM with the incorporation of data from relevant clinical studies as prior information, a dose of 600 mg BID (capsule) was determined for the dose-expansion phase. Capmatinib tablets were developed to improve patient convenience and compliance, and were determined to be the preferred formulation based on preliminary steady-state PK data from parallel clinical studies showing higher mean exposures [maximum serum concentration (Cmax) and area under curve (AUC) at steady-state] than the 600 mg capsules at the same dose levels tested, but within the range considering the coefficient of variability. 15 Overall, in the expansion stage of the study, 26/30 patients received capmatinib 600 mg BID in a capsule formulation and did not switch to the tablet form. Only two patients ongoing on 600 mg BID capsules switched to a tablet formulation and two patients were enrolled starting with capmatinib 400 mg BID tablet formulation.
Pharmacokinetics
PK analysis showed that capmatinib 300 mg BID capsules were rapidly absorbed with a median time to maximum plasma concentration (Tmax) of 2.0 h, a geomean Cmax of 2143.5 ng/ml, and a geomean AUC0−12 h of 7739.8 ng h/ml (cycle 1 day 1, Supplementary Table 2). The dose-normalized steady-state exposures of capmatinib were comparable with those from other capmatinib clinical trials. The exploratory analysis suggested that PK exposures were similar in patients with HCC, and patients with malignancies other than HCC, based on historical data from other clinical studies of capmatinib,15,17,18 and steady-state accumulation was minimal. The plasma concentration–time profile for capmatinib 300 mg BID capsules is presented in Supplementary Figure 1.
Safety
All patients (across dose-escalation and expansion phases) experienced at least one AE. The most common (>30%) AEs, regardless of causality, were nausea (42%), vomiting (37%), diarrhea (34%), aspartate transaminase (AST) increased, and decreased appetite (both 32%, Table 2). The most frequent (>5%) grade 3 or 4 AEs, regardless of causality, were AST increased (24%), blood bilirubin increased (16%), anemia (8%), and hyperbilirubinemia (8%, Table 2). The most common (⩾10%) AEs suspected to be study drug-related were nausea (39%), vomiting (32%), fatigue (21%), blood creatinine increased (13%), and diarrhea (11%, Supplementary Table 3). The majority of drug-related AEs were mild, and drug-related grade 3/4 AEs occurred in only five patients (13%, Supplementary Table 3). Serious AEs were reported in 19 (50%) patients and were most commonly abdominal pain, acute kidney injury, and esophageal varices (2 patients each). Only one patient had a serious AE (grade 3 vomiting) that was suspected of being related to study treatment. A total of nine patients (24%) had AEs that led to discontinuation of capmatinib, including two patients who had AEs suspected of being related to capmatinib [AST and alanine transaminase (ALT) increased, and amylase increased].
Adverse events [all grades (⩾10%) and grade 3/4] regardless of study drug relationship, by system organ class and maximum grade (safety set).
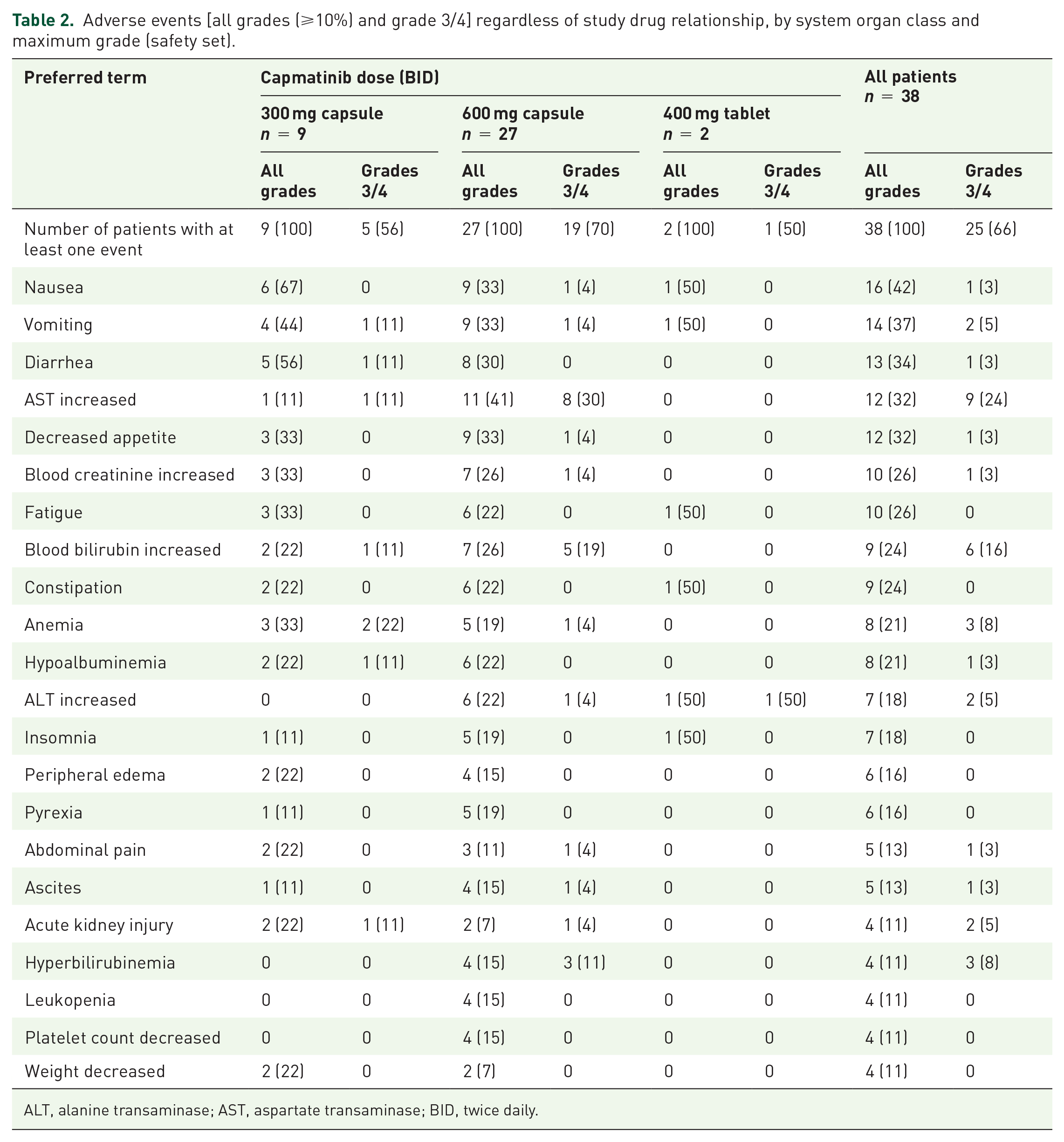
ALT, alanine transaminase; AST, aspartate transaminase; BID, twice daily.
Antitumor activity
Due to the limited number of MET-high patients enrolled in this study, a robust analysis of the primary endpoint of TTP was not possible. Therefore, objective responses were analyzed as a signal-seeking efficacy endpoint. In the dose-determining stage of the study (300 mg BID capsules, n = 8), stable disease was reported as the best overall response in two patients (25%, Table 3). In the expansion stage of the study, 20/30 patients had MET-low tumor expression (enrolled before the protocol amendment to revise the MET criteria) and 10/30 had MET-high tumor expression. At the data cut-off date, the overall response rate and disease control rate in all expansion patients (n = 30) was 10% (CI 95% 2.1−26.5) and 33% (CI 95% 17.3−52.8), respectively, regardless of MET status, all responders had MET-high status. In the MET-high expansion group (n = 10), the overall response rate was 30% (CI 95% 6.7−65.2) and the disease control rate was 50.0% (CI 95% 18.7−81.3), including 1 durable complete response (>600 days) and 2 partial responses [1 durable (>600 days) Figure 1]. In this study, 6/10 patients with MET-high status achieved tumor shrinkage (Figure 2). Of these, 3/6 patients showed significant tumor reductions, including 1 of 75% (this patient achieved a complete response) and another of 72% relative to baseline. The best percentage change from baseline in target lesions in the expansion stage of the study for MET-high and MET-low patients is shown in Figure 2.
Best overall response per RECIST v1.1 by study stage and MET status (full analysis set).
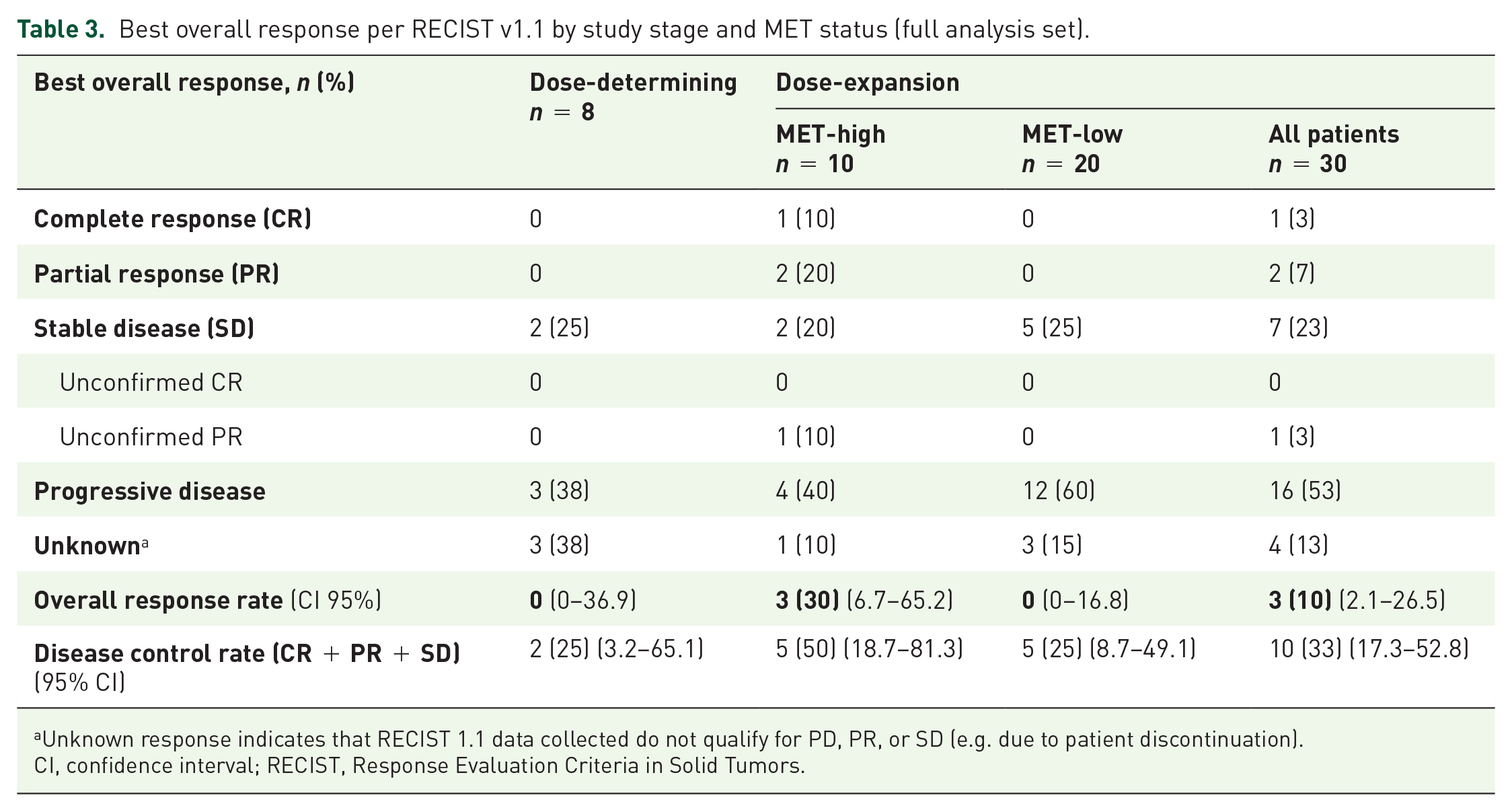
Unknown response indicates that RECIST 1.1 data collected do not qualify for PD, PR, or SD (e.g. due to patient discontinuation).
CI, confidence interval; RECIST, Response Evaluation Criteria in Solid Tumors.

Best percentage change from baseline in target lesions (RECIST v1.1; investigator assessed), dose-expansion stage (full analysis set).
Biomarker and sequencing analysis
Next-generation sequencing analysis of tumor biopsy samples from 10 patients was performed, and of these, 3 patients had MET-high tumors. These analyses revealed MCL1 GCN increases (CN 8−9) in 3/10 patients and MYC GCN increases (CN 8−13) in 2/10 patients, with co-amplification in 2 cases. Mutations in TP53 (short variants) were reported in 3/10 patients (Supplementary Table 4). Baseline serum HGF biomarker analysis from 35 patients showed no clear trend or correlation with MET status or clinical response in neither all patients nor in the MET-high group (Figures 2 and 3). In addition, there was no clear correlation of serum HGF level over time and clinical response in these patients (data not shown).

Best percentage change from baseline in tumor lesions by baseline serum HGF level.
Discussion
Currently, the standard-of-care first-line systemic therapy for patients with unresectable advanced HCC is the multiple receptor tyrosine kinase inhibitor sorafenib. Approval of sorafenib was based on two international, phase III studies. In a study in Caucasian patients, sorafenib provided an improvement in median overall survival [10.7 months versus 7.9 months for placebo (hazard ratio (HR) 0.69 in the sorafenib group)], disease control rate was 43% versus 32% for placebo, overall response rate was 2% versus 1% for placebo, and median TTP was 5.5 months versus 2.8 months for placebo. 19 In a parallel Asia-Pacific region phase III study, similar results were reported, with an improvement in median overall survival of 6.5 months for sorafenib, versus 4.2 months for placebo (HR 0.68 in sorafenib group). 20 The multiple receptor tyrosine kinase inhibitor (vascular endothelial growth factor receptors and others) regorafenib was FDA-approved for the treatment of patients progressing on or after sorafenib treatment, based on a phase III study that demonstrated an improvement in overall survival (10.6 months versus 7.8 months for placebo), the overall response rate was 11% versus 4% in the placebo arm. 21 Subsequently, the programmed death-1 (PD-1) inhibitor nivolumab received accelerated FDA approval as second-line therapy following progression on sorafenib, an overall response rate of 14% in sorafenib-treated patients and 20% in all patients was reported in the CheckMate 040 study, with the majority of patients with advanced HCC experiencing durable responses.22,23 Sorafenib remains the only approved first-line treatment for advanced HCC, and there is an urgent unmet need for alternative treatment options to be developed.
The heterogeneity of patient populations in most studies, in combination with a lack of patient selection according to the molecular signature, has led to other targetable oncogenic driver pathways being actively sought. Based on available data implicating the MET pathway in the tumorigenesis of HCC, and the poor prognosis for patients with the disease, the study described here was designed to evaluate the safety and efficacy of the MET inhibitor capmatinib in patients with advanced HCC, who have tumors meeting specific criteria for highly dysregulated MET signaling. In this study, encouraging antitumor activity was observed in the limited number of patients with advanced HCC and MET-high dysregulation status who were treated with capmatinib, with one complete response and two partial responses in patients in the MET-high status dose-expansion group (for an overall response rate of 30%). Only two of the responding patients (including a complete response) were on treatment for over 600 days at the time of the data cut-off date and were still on treatment with confirmed clinical benefit.
Overall, orally administered single-agent capmatinib 600 mg BID capsules and 400 mg BID tablets are tolerable with a manageable safety profile in patients with advanced HCC. No new safety findings were revealed, and the most frequent drug-related AEs were usually mild nausea, vomiting, and fatigue, and drug-related grade 3 or 4 AEs were uncommon.
The patient enrollment of this study was halted due to the difficulty in identifying patients who met the revised eligibility criteria of MET positivity and, therefore, the primary endpoint of TTP was not performed. Preliminary data based on a limited number of patients with MET-high HCC indicate that patients only responded to the treatment if they had a highly dysregulated MET pathway.15,17,18 Although overexpression or activation of MET has been reported in 20–48% of patients with HCC, based on a wide range of methods and cut-offs,4–8 these studies have not reported on the precise incidence and distribution of MET-high disease. In this study, our observation is that the incidence of MET-high disease is very low in previously untreated patients with HCC, with only 17/328 patients (5.2%) having tumors classified as MET-high after the second protocol amendment (defined as MET IHC intensity score 3+ in ⩾50% tumor cells, or 2+ in ⩾50% of tumor cells plus MET GCN ⩾5 by FISH, or MET GCN ⩾5 by FISH alone if IHC unavailable).
Clinical studies of other MET inhibitors in previously treated patients with HCC have employed different cut-off criteria for MET dysregulation/positivity, and have provided mixed results. In a phase Ib study carried out in Asian patients with advanced HCC, the MET inhibitor tepotinib had only limited antitumor activity in unselected patients, and 2/27 (7.4%) patients had partial responses, with a disease control rate of 37%. However, in that study patients were retrospectively evaluated for MET expression, which was defined as ⩾50% tumor IHC 2+/3+, and both responders had MET-positive tumors (2/7 MET-positive patients had partial responses).24,25 In contrast, in the negative METIV-HCC second-line phase III study (NCT01755767), tivantinib was evaluated in patients with MET-high HCC defined by IHC ⩾2+ in ⩾50% of tumor cells. 26 However, it should be noted that the extent of tivantinib’s clinical activity through MET inhibition has been questioned.27,28 However, these studies suggest that despite a reduced pool of eligible patients, stricter criteria are required for the selection of patients with MET-high HCC that are likely to benefit from MET inhibitor therapy.
Biomarker studies did not reveal any clear trends or correlations between serum HGF levels with clinical response. Next-generation sequencing of 306 cancer-related genes or gene rearrangements was performed with remaining biopsy material from 10 patients. However, only three of those patients were categorized as MET-high, precluding any correlative analysis with clinical response. BCL2-family apoptosis regulator MCL1 GCN increases were detected in 3/10 patients and MYC GCN increases were detected in 2/10 patients, with co-amplification in 2 cases. Both these genes encode proteins with roles in cell cycle progression, apoptosis, and cellular transformation, and have potential molecular and functional interactions with the MET pathway. MCL1 has also been reported as a possible response predictor for MET inhibition in patients with MET-high HCC. 29 In this study, only one tumor with MET-high status showed concomitant copy number increases in MYC and MCL1, but the response to treatment in this patient could not be determined because the patient discontinued treatment due to an AE. Mutations in TP53 (short variants) were also reported in 3/10 patients, mutant P53 has been implicated in the enhancement of MET trafficking, promoting MET recycling, and enhancing MET signaling. 30 However, in this study patient numbers were not large enough to draw any conclusions on the predictive significance of the genomic alterations that were observed.
Overall, the antitumor activity observed in this study in patients with MET-high tumors indicates that strict biomarker selection criteria, that were applied in this study, are required for the successful treatment of patients with HCC with single-agent MET inhibitor therapy, and that capmatinib represents a promising strategy for anti-MET therapy in appropriately selected patients with advanced MET-high HCC.
Supplemental Material
INC280X2201_HCC_Manuscript_Supplementary_Figure_S1 – Supplemental material for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma
Supplemental material, INC280X2201_HCC_Manuscript_Supplementary_Figure_S1 for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma by Shukui Qin, Stephen Lam Chan, Wattana Sukeepaisarnjaroen, Guohong Han, Su Pin Choo, Virote Sriuranpong, Hongming Pan, Thomas Yau, Yabing Guo, Minshan Chen, Zhenggang Ren, Jianming Xu, Chia-Jui Yen, Zhong-Zhe Lin, Luigi Manenti, Yi Gu, Yongjian Sun, Ralph Tiedt, Lu Hao, Wenjie Song and Tawesak Tanwandee in Therapeutic Advances in Medical Oncology
Supplemental Material
INC280X2201_HCC_Manuscript_Supplementary_Table_S1 – Supplemental material for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma
Supplemental material, INC280X2201_HCC_Manuscript_Supplementary_Table_S1 for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma by Shukui Qin, Stephen Lam Chan, Wattana Sukeepaisarnjaroen, Guohong Han, Su Pin Choo, Virote Sriuranpong, Hongming Pan, Thomas Yau, Yabing Guo, Minshan Chen, Zhenggang Ren, Jianming Xu, Chia-Jui Yen, Zhong-Zhe Lin, Luigi Manenti, Yi Gu, Yongjian Sun, Ralph Tiedt, Lu Hao, Wenjie Song and Tawesak Tanwandee in Therapeutic Advances in Medical Oncology
Supplemental Material
INC280X2201_HCC_Manuscript_Supplementary_Table_S2 – Supplemental material for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma
Supplemental material, INC280X2201_HCC_Manuscript_Supplementary_Table_S2 for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma by Shukui Qin, Stephen Lam Chan, Wattana Sukeepaisarnjaroen, Guohong Han, Su Pin Choo, Virote Sriuranpong, Hongming Pan, Thomas Yau, Yabing Guo, Minshan Chen, Zhenggang Ren, Jianming Xu, Chia-Jui Yen, Zhong-Zhe Lin, Luigi Manenti, Yi Gu, Yongjian Sun, Ralph Tiedt, Lu Hao, Wenjie Song and Tawesak Tanwandee in Therapeutic Advances in Medical Oncology
Supplemental Material
INC280X2201_HCC_Manuscript_Supplementary_Table_S3 – Supplemental material for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma
Supplemental material, INC280X2201_HCC_Manuscript_Supplementary_Table_S3 for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma by Shukui Qin, Stephen Lam Chan, Wattana Sukeepaisarnjaroen, Guohong Han, Su Pin Choo, Virote Sriuranpong, Hongming Pan, Thomas Yau, Yabing Guo, Minshan Chen, Zhenggang Ren, Jianming Xu, Chia-Jui Yen, Zhong-Zhe Lin, Luigi Manenti, Yi Gu, Yongjian Sun, Ralph Tiedt, Lu Hao, Wenjie Song and Tawesak Tanwandee in Therapeutic Advances in Medical Oncology
Supplemental Material
INC280X2201_HCC_Manuscript_Supplementary_Table_S4 – Supplemental material for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma
Supplemental material, INC280X2201_HCC_Manuscript_Supplementary_Table_S4 for A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma by Shukui Qin, Stephen Lam Chan, Wattana Sukeepaisarnjaroen, Guohong Han, Su Pin Choo, Virote Sriuranpong, Hongming Pan, Thomas Yau, Yabing Guo, Minshan Chen, Zhenggang Ren, Jianming Xu, Chia-Jui Yen, Zhong-Zhe Lin, Luigi Manenti, Yi Gu, Yongjian Sun, Ralph Tiedt, Lu Hao, Wenjie Song and Tawesak Tanwandee in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
Medical writing and editorial support for this manuscript was provided by Matthew Naylor PhD of Articulate Science and funded by Novartis Pharmaceuticals Corporation.
Author note
This study was presented in part at American Society of Clinical Oncology (ASCO), Chicago, IL, USA, 3–7 June 2016 (Poster Abstract 4074).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support for this study was provided by Novartis Pharmaceuticals Corporation.
Conflict of interest statement
Su Pin Choo received funding, nonfinancial support, and honoraria from BMS, nonfinancial support and honoraria from Bayer, and honoraria from Novartis, Shire, Sirtex, Eisai, and Celgene. Virote Sriuranpong has received research support from Novartis. Shukui Qin, Stephen Lam Chan, Wattana Sukeepaisarnjaroen, Guohong Han, Hongming Pan, Thomas Yau, Yabing Guo, Minshan Chen, Zhenggang Ren, Jianming Xu, Chia-Jui Yen, Zhong-Zhe Lin, and Tawesak Tanwandee have no competing financial interests. Yi Gu, Yongjian Sun, Lu Hao, and Wenjie Song are employees of Novartis. Luigi Manenti and Ralph Tiedt are employees of Novartis and hold stock with Novartis.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.