
Abstract
One of the mitogen-activated protein kinases (MAPKs), c-Jun NH2-terminal protein kinase (JNK) plays an important role in regulating cell fate, such as proliferation, differentiation, development, transformation, and apoptosis. Its activity is induced through the interaction of MAPK kinase kinases (MAP3Ks), MAPK kinases (MAP2Ks), and various scaffolding proteins. Because of the importance of the JNK cascade to intracellular bioactivity, many studies have been conducted to reveal its precise intracellular functions and mechanisms, but its regulatory mechanisms remain elusive. In this review, we discuss the molecular characterization, activation process, and physiological functions of mitogen-activated protein kinase kinase 7 (MKK7), the MAP2K that most specifically controls the activity of JNK. Understanding the role of MKK7/JNK signaling in physiological conditions could spark new hypotheses for targeted anticancer therapies.
Introduction
The mitogen-activated protein kinase (MAPK) pathway is a major cell-mediated cascade that regulates processes such as cell growth, differentiation, stress response, survival, and cell death in response to endogenous stimuli such as growth factors, hormones, cytokines, mitogens, and stress.1–3 So far, four kinds of MAPK signaling processes have been found: extracellular signal-regulated kinase (ERK) 1/2, c-Jun N-terminal kinase (JNK), p38, and ERK5. 4 These cascades constitute the ERK 1/2, JNK 1/2/3, p38 α/β/γ/δ, and ERK5 subfamilies of MAPK, respectively. These subfamilies respond to various extracellular stimuli, c-Fos, activating transcription factor-2, p53, ETS domain-containing protein-1, and c-Jun to regulate de novo gene expression and induction.5–8 Each subfamily generally comprises a signaling cascade consisting of MAPK kinase kinases (MAP3Ks), MAPK kinases (MAP2Ks), and MAPKs that are sequentially and selectively activated. So far, 20 MAP3Ks, 7 MAP2Ks, and 11 MAPKs have been identified.9,10 Among the MAPKs, the JNK cascade can be induced by environmental stresses such as heat shock, growth factor, and ultraviolet (UV) light.11,12 It regulates intracellular physiological functions such as cell death, growth, and differentiation. This process relies on activation through the serial phosphorylation of MAP3Ks (mixed-lineage protein kinase: MLK, apoptosis signal-regulating kinase, and transforming growth factor beta-activated kinase 1: TAK1), MAP2Ks (mitogen-activated protein kinase kinase 4: MKK4, and mitogen-activated protein kinase kinase 7: MKK7), and JNK (a MAPK).
Due to the importance of the JNK cascade to intracellular bioactivity, many studies have been conducted to elucidate its exact mechanisms.13–16 JNK activation relies on two upstream MAPKs with distinct JNK activation sites: tyrosine phosphorylation by MKK4 and threonine phosphorylation by MKK7. For instance, using genetically disrupted mouse embryonic fibroblasts (MEFs), it was found that axin-mediated JNK activation depends mainly on MKK7, and dishevelled-induced JNK activation depends almost equally on MKK4 and MKK7, whereas virus latent membrane protein-1-mediated JNK activation depends primarily on MKK4. 17 JNK activity against stress responses such as UV irradiation, heat, and osmotic changes is significantly inhibited in MKK4 and MKK7 gene-deficient embryonic stem cells and MEFs, which confirms that MKK4 and MKK7 contribute to JNK activation.18,19 In MKK7-deficient cells, the activation of JNK by inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1 was almost entirely lost, but it decreased only 50% in MKK4-deficient cells. 20 Therefore, MKK4 is required for optimal JNK activation, but MKK7 is essential for JNK activation by pro-inflammatory cytokines. These findings underline that the different MKKs needed for JNK activation depend on many factors, such as the stimulus, different expression levels of MKK4 and MKK7, scaffolds, and other cell-type-specific regulators. 21
Although various experiments using MKK4 and MKK7 deletions have been carried out to determine the activation and functional effects of JNK, further research is needed to fully elucidate how JNK regulates cell physiology. Herein, we review studies about the regulation of JNK signaling by MKK7, along with its relevance to cancer cell survival. We focus on MKK7 rather than MKK4 because MKK4 can also stimulate p38 MAPK activity, 22 which requires more exploration because of p38’s functional role in cell survival.23–26 We focus on MKK7, an essential JNK activator, as a way to understand the JNK cascade in more detail, which will be helpful to subsequent researchers of the JNK cascade.
Molecular characterization of MKK7
MKK7, also known as signal regulatory protein kinase 2 (SEK2) and c-Jun N-terminal kinase kinase 2 (JNKK2), was first cloned using murine mRNA by researchers at Massachusetts Medical School in 1997. Primers for MKK7 were designed based on the coding sequence of the Drosophila JNK activation factor hemipterous, which is 70% analogous in amino acid sequence to the MKK7 Kinase domain.27,28 After the initial cloning of MKK7, this gene was identified on chromosomes of various species, such as humans, rats, zebrafish, horses, and chickens.29–32 Exons in the MKK7 gene undergo alternative splicing at the RNA level to form MKK7 isoforms, which have modifications in the N- and C-termini. The N-terminal-modified isoforms are identified by the Greek alphabet, and the C-terminal-modified isoforms are identified by numbers. To date, four variants in humans, six variants in mice, and two variants in rats have been found.29,32–34
MKK7 consists of three domains: the D (docking) domain, the Kinase domain, and the DVD (domain for versatile docking) domain. The D domain of MKK7, present at residues 22–81 and containing F-X or F-F-X2-ψ-X-ψ motifs (where F, X, and ψ stand for positively charged, intervening, and hydrophobic residues, respectively), is an essential part of binding JNK (Figure 1A). The D domain is correlated with the binding affinity and activity of MKK7–JNK. 35 The Kinase domain, located at residues 120–380, involves a Ser–Xaa–Ala–Lys–Thr (S–X–A–K–T) kinase motif that is phosphorylated by upstream MKKKs. 36 The DVD domain, located at residues 377–400, plays an important role in the docking of upstream MAP3Ks such as MLKs, ASKs, TAKs, and LZK (leucine zipper-bearing kinase).37–39

Scheme for the MKK7-dependent JNK pathway. (A) Domain mapping of the MKK7 protein. (B) Schematic illustration of the MKK-dependent JNK pathway, which is activated by the phosphorylation of two residues in the kinase/catalytic domain (Ser and Thr for MKK7; Thr and Tyr for JNK).
The activation process for MKK7
MKK7 activity can be increased by either MKK7-autophosphorylation or phosphorylation of the Ser and Thr residues of the S-X-A-K-T motifs in the Kinase domain by upstream MAP3K1 (mitogen-activated protein kinase kinase kinase 1, MEKK1), MAP3K2 (MEKK2), or MAP3K11 (MLK3).40–42 Autophosphorylation occurs differently depending on the MKK7 isotype. For example, N-terminal-modified MKK7γ1 is phosphorylated only by MEKK1 without autophosphorylation, as opposed to MKK7β. 34 In addition to being specifically activated by upstream kinases, binding between the MKK7 D and DVD domains and a substrate can change the structure of MKK7 to phosphorylate the Ser and Thr residues of the Kinase domain motif. Those reactions lead to the sequential binding and dissociation of MAP3K/MKK7/JNK complexes, which thus affects the signal amplification of the JNK pathway (Figure 1B).43,44
In addition to direct physical interaction with substrates, MKK7 activity is regulated by interaction with scaffold proteins, which play an important role in the assembly of MAP3K/MKK7/JNK complexes. Scaffold proteins do not have a direct catalytic function but play a crucial role in controlling the binding duration and signal intensity of MAP3K/MAP2K/MAPK complexes in the MAPK pathway.45,46 One hypothesis suggests that scaffold proteins are promoted so that limited MAPKs can balance their signaling relative to the number of MAP3Ks in mammalian cells. 47 Several JNK signaling-related scaffold proteins have been identified and lead to cell proliferation, differentiation, and apoptosis. Those found so far are JNK-interacting protein (JIP) 1, JIP2, JNK/stress-activated protein kinase-associated protein 1 (JSAP1)/JIP3, JNK-associated leucine zipper protein (JLP)/JIP4, and the Plenty of SH3 (POSH) protein.48–50
JIP1 contains the JNK binding domain (JBD), SRC homology (SH3) domain, and the phosphotyrosine-binding (PTB) domain and affects JNK1 and JNK2 activation. 51 JIP1 generally interacts with MAP3Ks such as MEKK3, MLK, DLK (dual leucine zipper-retaining kinase), and histidine protein kinase (HPK1), but MKK7 is the only MAP2K that interacts with JIP1. Therefore, simultaneous expression of JIP1, MLK, and MKK7 in the JNK signaling pathway enhances JNK activation.45,52 JIP2 contains the same domains (JBD, PTB, SH) as JIP1 and is found in several human tissues, including the brain, prostate, ovary, and pancreas. It interacts with both p38 and JNK. Like JIP1, JIP2 and JIP3 also interact with MKK7 and are involved in the formation of JNK signaling complexes.48,53,54 JIP3, identified through yeast hybrid screening as a binding partner of JNK-1, contains a leucine zipper domain rather than the SH3 domain found in other JIPs. JIP3 interacts with various MAP3Ks, such as MEKK1, MLK3, and ASK1, and also with the MAP2Ks MKK4 and MKK7. Although JIP3 is known to be associated with JNK1, JNK2, and JNK3, it has the highest affinity for JNK3.48,49,55
The POSH protein has a specific Rac1 binding site that can bind to GTP-bound active Rac1. 50 It is also involved in regulating JNK signaling complexes of MAP3Ks/MAP2Ks/JNKs, similar to the JIPs. 56 When the expression of POSH was inhibited in PC12 cells, apoptosis was inhibited, 57 leading to the hypothesis that Rac1-mediated apoptosis occurs through an interaction with POSH. 58 Recently, the possibility of synergy between POSH and the JIPs has been discovered, and that new aspect is being studied.59–61 It has also been shown that POSH plays a crucial role in neural development in the early embryonic stage and plays a role in immune response by regulating T cell function.62–65
Arrestins have also been reported as scaffolds for MAPK activation, although the mechanism by which they assemble MAPKs into a signaling complex remains unexplored. It is known that all four vertebrate arrestins can interact with JNK3, MKK4, and ASK1, but only arrestin-3 can mediate JNK3 activation. 66 Initial studies demonstrated that only MKK4, not MKK7, could bind to arrestin-3, 67 but later studies indicated that arrestin-3 could also interact with MKK7 and promote JNK3α2 phosphorylation.21,68 Notably, arrestin-3 binds MKK7 with a lower affinity than it does MKK4. 21 Interestingly, JNK3α2 could both enhance the association between arrestin-3 and MKK4 and reduce arrestin-3 binding to MKK7. 21 That finding also demonstrates how cooperative regulation of JNK3α2 by MKK4/MKK7 could be determined by the concentration of arrestin-3 needed to induce JNK3α2 phosphorylation, emphasizing the concentration-dependence of the scaffold effect. 69 It is widely accepted that the formation of scaffold–kinase complexes contributes to the effective regulation of the specificity, efficiency, and amplitude of signal propagation. 70 One recent study used the metaphor of a conveyor belt mechanism for JNK3 activation by scaffold proteins. Thus, an active JNK3 molecule becomes an inactive JNK3 by helping to build an arrestin-3/MKK4/MKK7 complex that causes signal amplification. 68
Physiological roles of MKK7
According to several recent studies, MKK7 is involved in various biological responses through both JNK-dependent pathways and JNK-independent pathways. Here, we discuss the role of MKK7 in growth and development and the regulation of programmed cell death, paying particular attention to cancer cells.
Role of MKK7 in growth and development
MKK7 is reportedly essential for hepatocyte formation in embryonic development. The embryos of MKK7 knockout mice died between E11.5 and E13.5 due to immature hepatocyte formation. 71 Similarly, primary MKK7–/– hepatoblasts showed defective cell proliferation, and the expression of the cyclin-dependent kinase 2 kinase associated with the G2/M phase cell cycle was inhibited. 72 In addition, MKK7 has been shown to play an important role in molecular signaling for retinal development and retinal axonal damage. When retinal axonal damage occurred in MKK7-deficient mice, it caused optic nerve formation failure, irregular retinal axon trajectory, retinal thinning, retinal ganglion cell aggregation, and dendritic formation of dopaminergic amacrine cells. 73
Role of MKK7 in programmed cell death and tumorigenesis response
Programmed cell death, defined as apoptosis, autophagy, and programmed necrosis, plays an important role in the development and maintenance of tissue homeostasis by balancing normal cell survival and death. 74 JNK signaling pathways are associated with pro-apoptotic and anti-apoptotic processes in different cell types.75,76 Similarly, an MKK7-deficient model was used to show that MKK7 mediates apoptotic responses to a variety of stresses. For example, apoptosis was induced by stimulating mitochondrial antiviral signaling proteins (MAVS) in MKK7–/– MEF cells. Interestingly, the apoptosis induced by MAVS in MKK7–/– MEF cells was JNK2-dependent, not JNK1-dependent. 77
Tumorigenesis is defined as a complex and dynamic process of initiation, progression, and metastasis. 78 The JNK kinase signaling pathways have also been implicated in tumorigenesis 79 through their regulation of cell survival, proliferation, differentiation, and metastasis in cancer cells. Although the exact mechanisms by which the MKK7-JNK signaling axis regulates tumorigenesis remain to be elucidated, many studies have been conducted to clarify the relationship between MKK7 and diverse cancer cells.
In one recent study, five rare polymorphisms of MKK7 (p.Glu116Lys, p.Asn118Ser, p.Arg138Cys, p.Ala195Thr, and p.Leu259Phe) were analyzed in lung cancer patients. Among them, patients with the MKK7 p.Glu116Lys polymorphism had a significantly higher rate of lung cancer metastasis than the others. The p.Glu116Lys mutation affects the proliferation and metastasis of lung cancer cells by regulating a series of cancer-associated genes (upregulated: STC2, SLC1A3, MSMO1, BCL10, and HMGCR; downregulated: SAA1, SBK2, CDH5, COL4A2, and BCL9L). 80
Because the liver is larger than other organs and has an abundant blood supply, cancer cells often induce metastasis to the liver through blood. Thus, the prognosis of patients with advanced colorectal cancer varies greatly depending on the presence or absence of liver metastasis. In colorectal cancer patients, a potent inhibitor of hepatic metastasis was found, miR-493. The amount of miR-493 expressed and the incidence of metastatic cancer are closely related. In colorectal cancer patients, miR-493 inhibited the expression of MKK7 by targeting its 3’- untranslated region. Inhibiting MKK7 expression with miR-493 significantly reduced the liver metastases of colon cancer cells. In other words, the occurrence of liver metastases from primary colorectal tumors is associated with elevated MKK7 levels. 81
T-cell acute lymphoblastic leukemia (T-ALL) is a type of aggressive acute leukemia with a high recurrence rate. The incidence of T-ALL is about 15% of pediatric cases and 25% of adult cases. 82 Although the incidence of pediatric T-ALL is lower than in adults, the recurrence rate of pediatric T-ALL is higher than that of adult T-ALL. In pediatric lymphoblastic leukemia, the gene for Kruppel-like factor 4 (KLF4), referred to as a zinc-finger transcription factor, is inhibited by DNA methylation. 83 The loss of KLF4 in leukemic cells accelerated the development of T-ALL by enhancing the G1 to S phase transition. It is generally known that KLF4 regulates the JNK pathway by inhibiting the coding gene for MKK7. The absence of KLF4 in leukemic cells induces excessive MKK7 expression, which increases the proliferation of leukemic cells and eventually leads to T-ALL. 84
Thus, MKK7 expression induces the differentiation and metastasis of cancer cells, thereby promoting tumor progression. However, it also plays a role in reducing the proliferation of cancer cells through functions such as apoptosis. For example, in hepatocellular carcinoma (HCC), the level of TIP41-like protein (TIPRL) correlates directly with the level of apoptosis, so high levels of TIPRL inhibit the expansion of cancer growth.85,86 TIPRL is a negative regulator of protein phosphatase 2A (PP2A), a serine/threonine phosphatase targeting the Raf, MEK, and protein kinase B signaling systems.87–90 In HCC, TIPRL and MKK7 competitively bind to PP2A to regulate cell apoptosis. 86 In other words, PP2A binds to MKK7 to suppress MKK7 activity due to phosphorylation, thereby inhibiting the apoptosis reaction and promoting the proliferation of HCC. In another case, inactivation of MKK7 in KRasG12D-driven lung cancer increased tumorigenesis and reduced overall survival. 79 That response was due to the abnormal role of p53, which is essential for tumor development and cell cycle arrest.91,92 Because the stability of p53 is obtained through the MKK7-mediated JNK pathway, the loss of MKK7 activity disrupts the stability of p53, so MKK7 is not acting directly as a cancer suppressant. 79
Likewise, the ubiquitination-like post-translational neddylation of MKK7 in breast cancer cells inhibited its activity and positively affected cell proliferation. 93 A direct association between MKK7 and a fragment of RAN-binding protein 2 (RanBP2), recognized as SUMO E3 ligase, was confirmed.93–95 RanBP2 knockdown inhibition of MKK7 neddylation in breast cancer cells maintained MKK7 activity, which reduced the proliferation of human breast cancer cells and impaired the epithelial–mesenchymal transition. Furthermore, the phenomena associated with RanBP2 knockdown were reduced by concomitant MKK7 knockdown. 93
Regulatory functions of MKK7 in noncancerous cells have also been reported. Studies of primary MKK7–/– hepatoblasts, which have defective cell proliferation, suggest that the MKK7 signaling pathway is involved in cell proliferation through the regulation of Cdc2 expression. 72 In contrast, MKK7–/– mast cells showed hyperproliferation of IL-3 and stem cell factor through the upregulation of cyclin D1 caused by decreased expression of JunB and the cell cycle inhibitor p16INK4a. 30
MKK7 as a therapeutic target
In this section, we discuss several drug candidates that target MKK7, their modes of action, and their physiological relevance. The two main ways to selectively target MKK7 are developing a selective covalent inhibitor of MKK7 and targeting a specific protein–protein interaction of MKK7.
Covalent inhibition of MKK7
To date, selective and potent inhibitors of MKK7 have been poorly studied. Adequate selectivity is difficult to achieve because of the high structural homology among the MAP2Ks, particularly at the adenosine triphosphate (ATP) binding site, which is a common target binding region for kinase inhibitors. 96 Thus, designing and optimizing an inhibitor of a single kinase at allosteric sites is challenging, although several strategies are available to enhance selectivity across the kinome, such as targeting poorly conserved residues. Research efforts to identify selective MKK7 inhibitors began when it was found that 5Z4Z-7-oxozeaenol (5Z7O), an irreversible inhibitor that covalently links to the cysteine residue at the gatekeeper-2 position (a residue adjacent to the DFG-motif in ERK, TAK1, and MAP2K1), could bind to the ATP sites of MKK7 in a mode that differs from the binding mode of ERK, TAK1, and MAP2K1. 97 The unprecedented binding was possible because 5Z7O can covalently bind the Cys218 of MKK7, a residue that is not present in other MAP2Ks at a similar position, 97 making it a distinct and advantageous point for developing selectivity across the kinome. More recently, it was discovered that the unprecedented binding to Cys218 is crucial for the auto-inhibition form of MKK7. 96
Shraga et al. 98 systematically identified covalent inhibitors of MKK7 by covalently targeting this non-conserved cysteine using covalent docking, followed by hit optimization and multiple validations, such as genetic validation of on-target activity, assessment of its selectivity across the kinome and proteome, analysis of metabolic stability and, finally, trial on primary mouse B cells. The three best candidate inhibitors of MKK7 were (as indicated in the original manuscript) MKK7-COV-7, MKK7-COV-9, and MKK7-COV-12 (Figure 2). 98 These three candidates inhibited around 90% of the response of primary B cells to lipopolysaccharide, which is a level similar to that of JNK-IN-8, a potent and specific JNK inhibitor. 98 Although this interesting research has produced potential candidates for selective MKK7 inhibition and provided a starting point for further development, further validation and studies on in vivo systems are needed before any of the candidates can be used therapeutically.
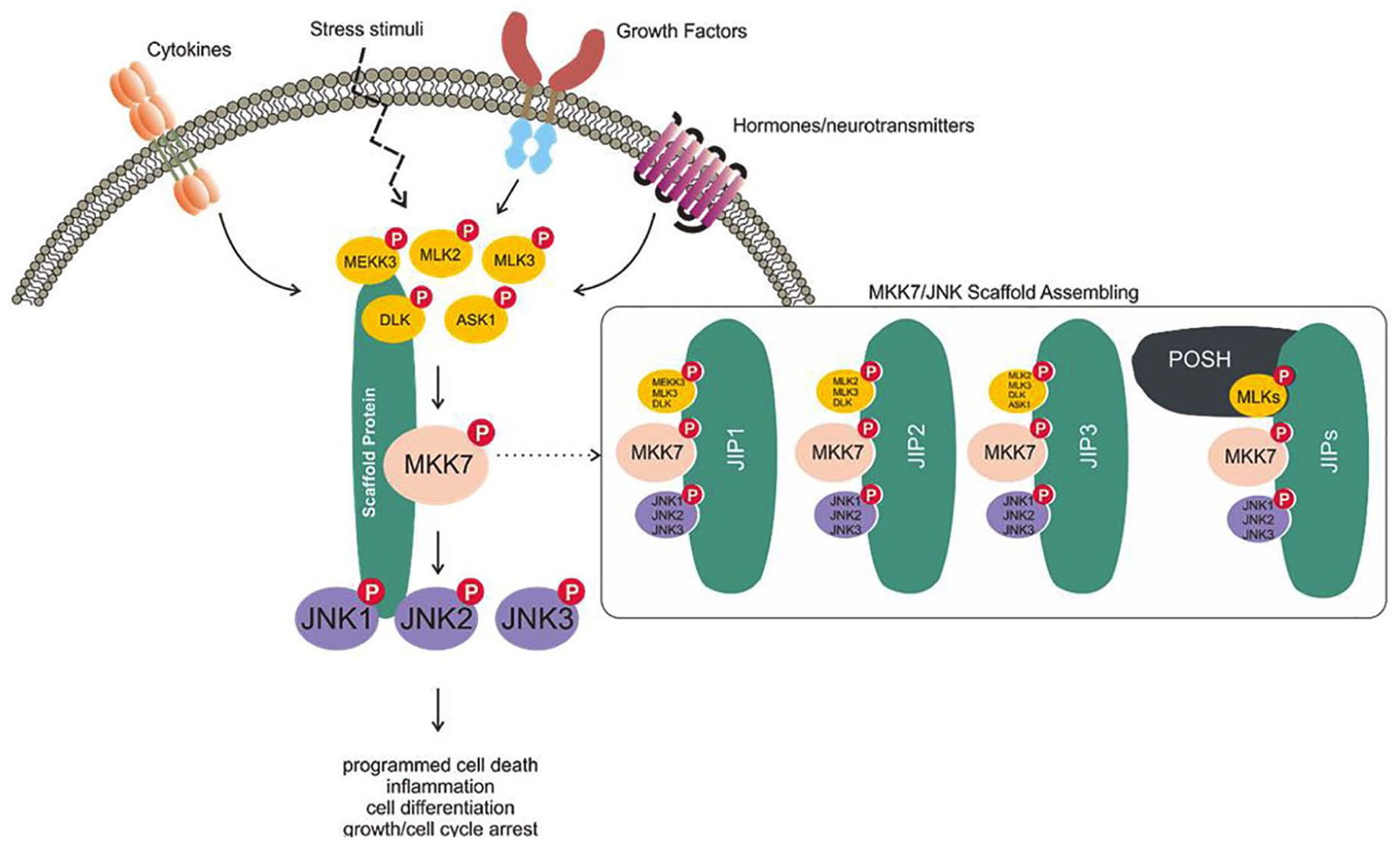
Putative scheme for the MKK7/JNK signaling pathway. The MKK7/JNK signaling axis can be triggered by cytokines, stress stimuli, growth factors, and hormones/neurotransmitters. This stimulation promotes the incorporation of an MKK7/JNK scaffold that mediates the phosphorylation of MAP3Ks (such as MEKK3, MLK2, MLK3, DLK, and ASK1), phosphorylates MKK7, and then phosphorylates the JNK kinases. These signaling cascades have been implicated in regulating various physiological functions, including programmed cell death, inflammation, cell differentiation, and growth/cell cycle arrest.
Targeting an MKK7 binding partner (protein–protein interactions)
Targeting a specific protein–protein interaction of MKK7 might resolve the risk of non-selective inhibition or the toxicity common to ATP analogue inhibitors, and compounds targeting the protein–protein interactions of MKK7 have indeed been reported. Growth arrest and DNA damage-inducible beta (GADD45β), a small acidic protein whose expression denotes aggressive disease in multiple myeloma (MM), 99 has been reported to physically interact with MKK7. 100 Enzymatic inhibition of MKK7 happens when GADD45β is able to interfere with the access of ATP to the catalytic pocket of MKK7. Tornatore et al. 99 have identified a peptide-based structure, D-tripeptide (DTP3) (Figure 3), that can specifically target the GADD45β–MKK7 interaction. DTP3 works by binding to MKK7 with a high affinity, producing a conformational change that facilitates the displacement of GADD45β.99,100 The interface models between the GADD45β–MKK7 and MKK7–DTP3 complexes demonstrate that the interactions of GADD45β and DTP3 with MKK7 are mutually exclusive. As reported by Rega et al. 100 ‘DTP3 interacts with two spatially adjacent outer MKK7 region that form a shallow pocket located proximally to the ATP pocket; while in the presence of GADD45β, the DTP3-binding region is partly occupied by loop 2 of the GADD45β.’

Chemical structure of compounds targeting MKK7/JNK-signaling.
Regarding its therapeutic relevance, constitutive NF-κB signaling promotes survival in cancers, including MM. 99 Therefore, a therapeutic strategy that can selectively target NF-κB is highly desired. One way to achieve such specificity is by targeting NF-κB target genes that contribute to the anti-apoptotic mechanism of cancer cells. GADD45β, a transcriptional target of NF-κB, also mediates MKK7/JNK signaling inhibition, which contributes to survival in MM conditions. 99 MKK7 is an upstream activator of pro-apoptotic JNK kinases, so inhibition of this signaling axis with a GADD45β interaction can also inhibit apoptosis.99,101,102 DTP3 has a high therapeutic index in vitro and displays potent activity against MM in vivo by selectively inhibiting the NF-κB survival pathway for MM. 99 The current understanding of DTP3 as a selective anticancer agent is limited to MM; therefore, further research should be conducted in other types of cancer or other pathological conditions associated with aberrant expression of GADD45β, such as several types of lymphoma, HCC, 103 pituitary gonadotrope tumors, 104 and colorectal cancer.105,106
Using a similar mechanism, cellular caspase 8 (FLICE)-like inhibitory protein (c-FLIPL) can also interact directly with MKK7, disrupting the interactions between MKK7 and MAP3Ks and inhibiting prolonged activation of the MKK7-JNK pathway. 107 Despite reports that some chemical agents can alter c-FLIP expression in ways relevant to cancer therapy, 108 selective and potent agents that specifically target the cFLIPL–MKK7 interaction remain to be elucidated. Moreover, the correlation and coordination of cFLIPL and GADD45β in controlling MKK7 remain largely unknown. Cordycepin has been showed to inhibit TNF-α-mediated NF-κB/GADD45 signaling by upregulating MKK7-JNK signaling activation through the inhibition of c-FLIPL expression, 109 although the target specificity still needs to be validated.
The TOR signaling pathway regulator-like protein contributes to TNF-related apoptosis-inducing ligand (TRAIL) resistance by forming an MKK7-PP2Ac-TIPRL complex.110,111 Binding of TIPRL to MKK7 and PP2Ac restricts the prolonged activation of MKK7 by tethering the PP2Ac phosphatase, which in turn facilitates MKK7 dephosphorylation, suppressing the JNK/caspase axis and inhibiting TRAIL-induced cell death in TRAIL-resistant cancer. 111 Yoon et al. 110 elucidated MKK7-TIPRL interaction inhibitors using high-throughput enzyme-linked immunosorbent assay screening followed by hit-to-lead optimization, and they reported two promising compounds, TRT-0029 and TRT-0173 (as designated in the original manuscript) (Figure 3). These indazole-based compounds act as TRAIL sensitizers. In a combination treatment with TRAIL, these compounds enhanced TRAIL-induced apoptosis in in vitro systems of Huh7 cells and suppressed tumor growth in vivo in a mouse xenograft model. 110
Other protein binding partners worth mentioning are the receptor for activated C kinase 1 (RACK1), the small-GTPase Rac1, and Ras-association domain family 7 (RASSF7). RACK1 has been implicated in regulating the activity of the JNK pathway. Although the molecular mechanism by which RACK1 regulates the JNK pathway could be cell context-dependent, it has been reported that RACK1 can interact directly with MKK7 in in vitro and in vivo systems, aiding the binding of MKK7 to MAP3Ks, which in turn enhances MKK7/JNK activity in HCC. 112 Rac1 is involved in the activation of the MKK7-JNK signaling pathway, which induces Atg5 expression and consequently autophagic cell death in response to oncogenic H-ras. 113 Meanwhile, RASSF7 associates specifically with the phosphorylated form of MKK7, maintaining MKK7’s phosphorylated state even in the absence of stress stimuli (independent of MAP3Ks activation). 114 Takahashi et al. 114 proposed that RASSF7 could impose a rigid interaction with phosphorylated MKK7 that would likely cause a decrease in phospho-MKK7’s ability to contact subsequent substrates, such as JNK or a phosphatase. As a result, the RASSF7–MKK7 interaction contributes to anti-apoptotic regulation by inhibiting JNK phosphorylation. 114 As RASSF7 and phospho-MKK7 accumulate (which can also be accelerated by stress stimuli), RASSF7 tends to degrade, and subsequent JNK-mediated apoptosis can proceed. 114 Growing evidence demonstrates the important role of this protein in both tumorigenesis and cell death responses. Chemical agents that regulate Rac1 with respect to cancer angiogenesis and metastasis have been reported elsewhere, 115 but a chemical agent with a selective mechanism targeting Rac1/MKK7/JNK has not been found. Furthermore, no studies have reported chemical compounds that regulate the interaction between RACK1 or RASSF7 and MKK7.
Compelling evidence suggests that this signaling axis is crucial to tumorigenesis. MKK7/JNK axis signaling has been intriguingly implicated in both positively and negatively regulating apoptosis and other types of cell death, independent of the cellular context. Therefore, a better understanding of cellular-context specificity and subsequent regulatory mechanisms is still needed, along with a consideration of the intricate interactions between MKK7 and various cascade components. Some selective inhibitors of this signaling axis have been elucidated, along with their modes of action (Figure 4); nevertheless, chemical agents targeting MKK7-binding partner proteins could be an option for providing specificity and delineating context-dependent regulation of JNK signaling. Because JNK signaling is not limited to regulating apoptosis/tumorigenesis, confirmatory studies of these candidate inhibitors in multiple pathological conditions must be included in future studies. Based on our observations here, several potential targets for MKK7/JNK signaling have not been adequately studied. Therefore, many paths are open for future studies.

Mode of action of selective chemical agents targeting MKK7. (*) need to be further validated for selectivity.
Conclusion
The JNK cascade is a major MAPK signaling response to many extracellular stimuli. JNK cascades can be activated transiently or continuously by various kinases, scaffolding proteins, and phosphatases. Of the kinases, MKK7 is the MAP2K most essential to regulating the activity of the JNK cascade. Many in vitro and in vivo studies have reported the importance of MKK7 to diverse intracellular functions, such as cell growth, proliferation, senescence, differentiation, transformation, cell cycle regulation, and tumor metabolism. The loss of MKK7 function in cells and mice disrupts many key processes needed to maintain organic homeostasis, such as apoptosis, cell formation and development, and tumorigenesis. However, it is difficult to judge the exact role of MKK7 because its physiological and pathological functions are highly contradictory.
Therefore, further studies are needed to elucidate the detailed molecular mechanisms of various scaffold proteins that interact with MKK7 to regulate cell survival and proliferation. Defining the role of MKK7 in tumorigenesis will have a profound effect on future cancer prevention and treatment strategies.
Footnotes
Author contributions
Jae Gwang Park, Nur Aziz, and Jae Youl Cho designed the study, interpreted the data, and wrote and revised the manuscript.
Author note
Jae Gwang Park is also affiliated to Division of Translational Science, Research Institute, National Cancer Center, Goyang 10408, Republic of Korea.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by BK21 PLUS and the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2017R1A6A1A03015642), Korea.
Conflict of interest statement
The authors declare that there is no conflict of interest.