
Abstract
A coordinated action of innate and adaptive immune responses is required to efficiently combat a microbial infection. It has now become clear that cancer therapies also largely benefit when both arms of the immune response are engaged. In this review, we will briefly describe the current knowledge of innate immunity and how this can be utilized to prime tumors for a better response to immune checkpoint inhibitors. Comments on compounds in development and ongoing clinical trials will be provided.
Introduction
Recent advances in cancer immunotherapy have been achieved with antibodies that inhibit immune checkpoint receptors on immune cells and tumor cells, mainly programmed-cell death 1 (PD-1), its ligand PD-L1 and cytotoxic T lymphocyte-associated molecule-4 (CTLA-4). 1 However, most cancer patients do not respond to single checkpoint inhibition.2–4 Among the reasons is the lack of tumor infiltration by cytotoxic CD8+ T-cells. 5 Non-T-cell-infiltrated tumors (‘cold tumors’) probably require the combination of checkpoint inhibitors with other therapies designed to attract these effector cells into the tumor microenvironment.
The immune system has two arms, (i) the innate immune arm that is rapidly activated after an appropriate stimulus but lacks antigen-specificity and memory, and (ii) the adaptive immune response that requires time to appear but is antigen-specific and long-lasting. 6 Both arms of the immune system are intimately linked such that the innate arm provides the conditions for an efficient activation of the adaptive response (Figure 1).

From stimulating innate immune responses to an adaptive antitumor response. A schematic view is given. In the tumor microenvironment, activation of PRRs is achieved by the recognition of DAMPs released by dying tumor cells and by drugs such as TLR agonists, STING agonists, DNA demethylating agents and treatments based on the use of modified oncolytic viruses. The activation of the innate immune response leads to an efficient priming by dendritic cells of T-cells in lymph nodes and to the infiltration by tumor-specific T-cells into the tumor. Details are described in the text.
Elements of the innate defense system are physical barriers, soluble factors such as complement proteins, interferons (IFNs) and IFN-stimulated proteins, and immune cells such as dendritic cells, macrophages, neutrophils and natural killer (NK) cells. The adaptive arm of the immune system comprises T and B lymphocyte subpopulations that recognize pathogens in an antigen-specific way via divergent T-cell receptors and B-cell receptors, respectively. Among the latter are CD4+ T-helper and T-regulatory cells (Tregs), cytotoxic CD8+ T-cells, B-cells and antibody-producing plasma cells. 6
Several virus infections including Papilloma viruses, Merkel cell polyomavirus, and hepatitis B and C viruses can induce tumors in humans. Interestingly, such tumors that are linked to virus infections seem to respond better to checkpoint inhibitors than tumors that are not virus-linked.7,8 The reason for this seems related to a more activated innate immune response.9–16 Thus, like in virus infections, the innate immune system probably provides a better microenvironment for the development of a potent specific antitumor response.17,18
Type I IFNs are key regulatory elements in this aspect. They are produced for example when virus components or cell-derived damage-associated molecular patterns (DAMPs) bind and activate pattern recognition receptors (PRRs). 19 The secreted IFNs can then activate dendritic cells (DCs) in tumor-draining lymph nodes and enhance the cross-presentation of tumor-associated antigens to CD8+ T-cells, 20 which subsequently may lead to tumor-specific CD8+ T-cell expansion and tumor destruction.
A detailed understanding of the innate immune response against viruses may provide opportunities for developing more efficient treatments in the field of cancer immunotherapy. Indeed, the combination of checkpoint inhibitors with agents that trigger the innate immune response enhances their antitumor effect. Here, we will review the recently described means of activating innate immune responses to improve immunotherapy for cancer patients. Novel strategies that activate directly or indirectly PRRs will be commented on.
Pathogen recognition receptors (PRRs)
The innate immune response initiates with the recognition of foreign nucleic acids or other molecules in host cells by PRRs. PRRs recognize molecules derived from pathogen-associated molecular patterns, as well as DAMPs released from endogenous tissues that have suffered some damage. 21 The understanding of the immunostimulatory as well as pro- or antitumoral function of PRRs is necessary to exploit them for enhancing cancer immunotherapy.
Several different subtypes of PRRs are described today: Toll-like receptors (TLRs), NOD-like receptors (NLRs), c-type lectin receptors (CLRs), cytosol dsDNA sensors (CDSs) and retinol acid inducible gene 1 (RIG-1)-like receptors (RLRs).22,23 PRRs are classified according to their cellular location. They are located in cell membranes, such as TLRs, or in the cytoplasm like NLRs, CLRs, CDSs and RIG-1-like receptors (RLRs). 21 PRR activation induces the production of type I IFNs (mainly IFN-α proteins and IFN-β). 22 Subsequently, type I IFNs control the transcription of genes that are restricting viral infections (so-called ‘virus restriction factors’). In addition, type I IFNs activate NK cells, promote antigen presentation 24 and participate in the differentiation of specific CD8+ cytotoxic T lymphocytes (CTLs). Finally, type I IFNs have antiproliferative functions that are through TP53 gene induction.25,26
TLRs constitute a receptor family that is mainly expressed on macrophages and DCs. In humans, the family has 11 members 27 located on the extracellular membrane (TLR 1, 2, 4, 5, 6 and 11) or in the intracellular counterpart of endosomes (TLR 2, 3, 7, 8, 9 and 10). Cell membrane-bound TLRs recognize glycoproteins, while endosome-placed TLRs respond to nucleic acid molecules, in particular viral RNA.27–29
TLR4 was the first TLR identified. Activation of TLR4 signaling is preceded by binding of lipopolysaccharides produced by Gram-negative bacteria.30,31 In cancer, TLR4 activation has a dual role. Although its upregulation is associated with chemoresistance, 32 metastasis and immunosuppression 33 in several tumor types, TLR4 activation has also an anticancer effect. While TLR4 antagonists could help reduce metastasis, TLR4 agonists have been shown to induce antitumor immunity in patients and models of cancer. Several TLR4 agonists, such as OM-174, 34 or the Streptococcus-derived agent OK-432,35,36 Coley toxin (a mixture of killed Streptococcus pyogenes and Serratiamarcescens bacteria) and Bacillus Calmette-Guerin, have antitumoral effects.36–38
Double-stranded RNA (dsRNA) is detected via TLR3 and RLRs (RIG1 and MDA5). Among immune cells, myeloid DCs and macrophages express TLR3. TLR3 is also expressed in fibroblasts and hepatocytes. When activated, TLR3, through TIR-domain-containing adapter-inducing interferon-β (TRIF), tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) and tankyrase 1 (TANK1), activates the transcription factors interferon response factor 3 (IRF-3) and nuclear factor kappa B (NF-κB). This then leads to the expression of type I IFNs, mainly IFN-β 39 (Figure 2). RLRs use protein adaptor mitochondrial antiviral signaling to activate IRF-3, IRF-7 and NF-κB 40 (Figure 2). RLRs are expressed in many tissues and play a prominent role in myeloid cells, fibroblasts, hepatocytes and central nervous system cells. While highly expressed in plasmacytoid DCs, RLRs are not essential for IFN type I production by these cells. 41
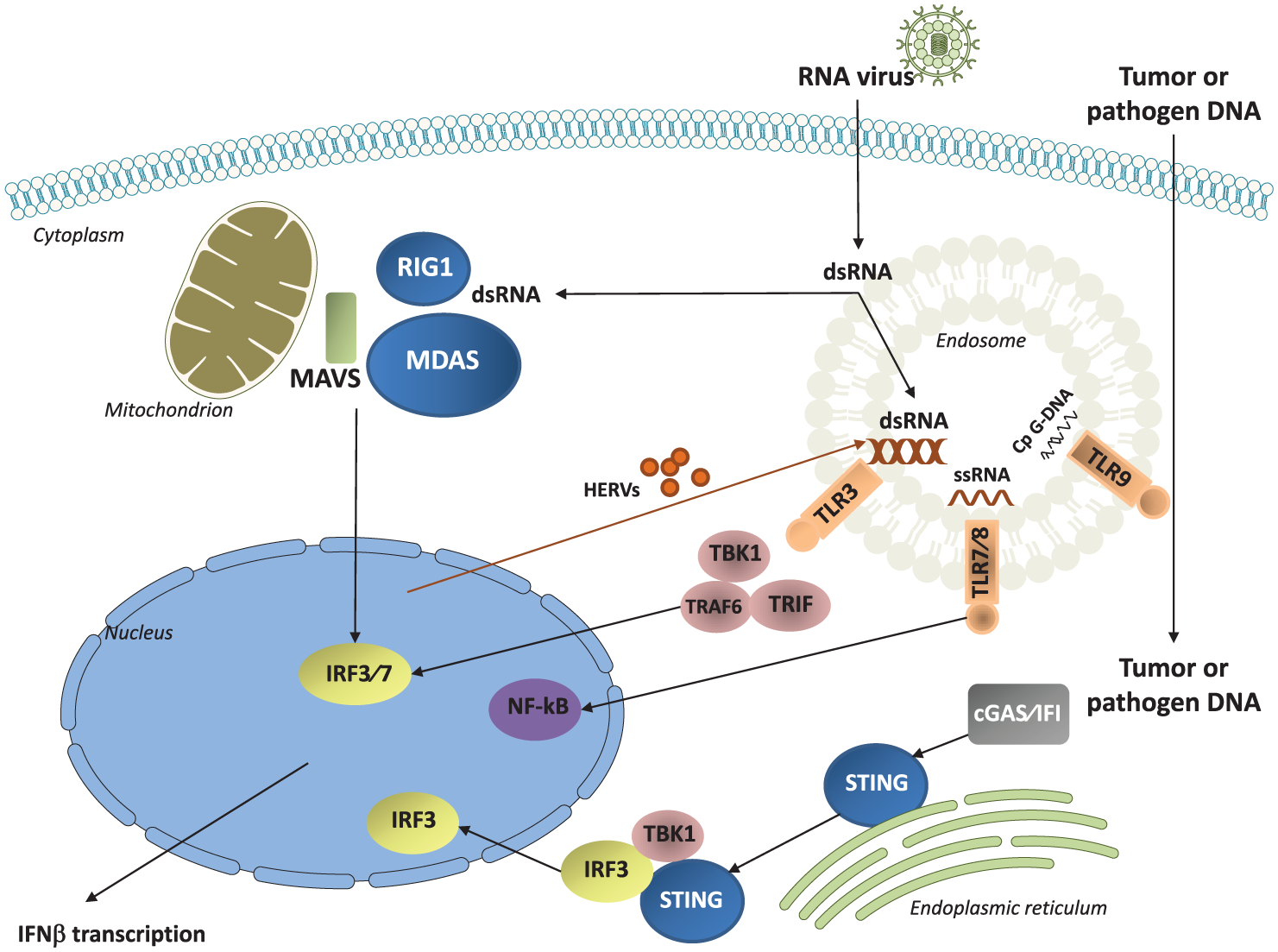
Main subtypes of PRRs as targets for cancer treatment: Toll-like receptors (TLR3, TLR7/8, TLR9), cytosol dsDNA sensors (cGAS/IFI that activate STING) and the retinol acid inducible gene RIG-1 like receptors (RIG1 and MDA5). Activation of these receptors, following detection of nucleic acids from virus, induces production of type I IFNs (IFN-α and IFN-β). dsRNA (from HERVs re-expressed after treatment with azacytidine or from exogenous infection) activates TLR3, MDA5 and RIG1; ssRNA activates TLR7/8; DNA (from pathogens or from tumor cells) activates STING.
The synthetic dsRNA BO-112 that activates melanoma differentiation-associated protein 5 (MDA5) demonstrated tumor-specific immune responses with a good toxicity profile in a first in-human trial. 42 A phase I clinical trial testing the combination of BO-112 with anti-PD-1 antibodies is ongoing in several centers of Spain (Table 1). Other interesting antitumoral compounds in development are the synthetic analogs of dsRNA polyinosinic:polycytidylic acid (poly I:C) that activate TLR337,43–45 and RIG1/MDA5 46 and 5′ triphosphate small interfering RNA (ppp-siRNA) that activates RIG1 and silences specific oncogenes like BCL-2 via RNA-interference. Both types of compounds have demonstrated antitumor activities in vivo. 47
Cancer clinical trials in progress with drugs targeting innate immune response.

CTLA-4, cytotoxic T lymphocyte-associated molecule-4; DNMT, DNA methyltransferase; HDAC, histone deacetylase; MDA, melanoma differentiation-associated protein; MDS, myelodysplastic syndromes; NCT, National Clinical Trials; NSCLC, non-small cell lung cancer; PD-1, programmed-cell death 1; PRR, pathogen recognition receptor; SCLC, small cell lung cancer; STING, stimulator of interferon genes complex; TLR, Toll-like receptor.
The detection of single stranded RNA (ssRNA) is due to TLR7 and TLR8 which activate NF-κB, IRF-3 and IRF-7, and lead to the expression of type I IFN, TNF-α, interleukin (IL)-1 and IL-12 48 (Figure 2). TLR7 is mainly expressed in plasmacytoid DCs and B-cells, while TLR8 is expressed in myeloid DCs and Tregs. Imidazoquinolinamin derivates, such as imiquimod, approved for the treatment of basal cell carcinoma, show antitumoral effects through TLR7 and TLR8 activation. 49 Intratumoral resiquimod (R848), a ligand of TLR7 and TLR8, in combination with anti-OX40 in animal models induces a systemic antitumor effect. 50 MEDI9197 (formerly 3M-052), is a novel TLR7/8 dual agonist formulated for intratumoral injection that has been studied in patients with cutaneous tumors, demonstrating safety and immunogenicity. 51
TLR9 recognizes unmethylated CpG motifs from bacteria and viruses 52 and RNA:DNA hybrids. 53 Its triggering activates plasmacytoid DCs and B-cells. 54 Several synthetic CpG oligonucleotides have been developed as TLR9 agonists mimicking natural CpG motifs (Table 1). The combination of intratumoral CpG SD-101 with anti-OX40, which triggers a T-cell immune response, demonstrated complete responses and long-term survival in animal models. 50 Similarly, data from a phase I clinical trial in advanced melanoma showed objective responses in four of the five patients treated with the combination of intratumoral CpG SD-101 and the anti-PD-1 antibody pembrolizumab. 55
The CDSs cGAS and IFI16 operate via stimulator of interferon genes complex (STING) to detect free DNA in the cytosol. When cytosolic DNA is detected by cGAS it catalyzes the STING ligand cGAMP. 56 STING is then activated, translocates to perinuclear endosomes and recruits TBK1 and IRF3. These molecules are phosphorylated and translocate to the nucleus with subsequent transcription of type I IFNs 56 (Figure 2). STING is essential for the production of type I IFN signaling in DCs which enables them to present tumor antigens and prime CD8+ T lymphocytes leading to T-cell infiltration into the tumor.18,57 STING is located at the cytosolic site of the endoplasmic reticulum membrane 56 and is activated by DNA from damaged tumor cells that reach the cytosol of DCs. 58 Some intratumoral-injected compounds can activate STING in mice, like 5,6-dimethylxanthenone-4-acetic acid (DMXAA). 59 In humans, the STING agonist ML RR-S2 CDA (MIW815, ADU-S100) is in clinical development 60 (Table 1).
Interferons
IFNs were discovered in the late 1950s during replication studies of influenza virus. 61 They are cytokines that play a critical role in innate immune responses against viral infections and participate in the activation of the adaptive immune response. 26 Several types of IFNs have been described including type I IFNs (various IFN-α, IFN-β, and others), the type II IFNγ, and the more recently classified type III IFNs (IFN-λ). Type I IFNs are produced by most cell types: fibroblasts, DCs and hepatocytes (through RIG-1), plasmacytoid DCs (through TLR9 and TLR7/8) and macrophages and hepatocytes (through TLR3 and TLR4). Type II IFNs are mainly secreted by T-helper-1 (Th-1) lymphocytes, CD8+ lymphocytes, NK and NK T-cells. 62
All the different types of IFNs signal through IFN receptors that are differently distributed among cell types. IFN-β binds to the IFN-α/β receptor (IFNAR) to further activate production of more type I IFN 63 (Figure 3). IFNAR activates janus kinase (JAK) family members JAK1 and Tyk-2, and subsequently signal transducer and activator of transcription 1 (STAT1) and 2 (STAT2). STAT1/2 bind to IRF9 (p48) and form the IFN-stimulated gene factor 3 (ISGF3; Figure 3). ISGF3 initiates the transcription of several interferon-stimulated genes (ISGs) by binding to the promoter region of IFN-stimulated response elements (ISRE; Figure 3). ISGs include PKR, IRF-1 and IRF7. When activated by TBK-1/IKKe, ISGs regulate IFNα gene transcription. 64 ISGs activate antimicrobial programs that both degrade viral proteins and inhibit cancer cell proliferation 65 (Figure 3). Type I IFNs also stimulate the adaptive immune response. Specifically, they promote major histocompatibility complex (MHC) class I and II expression on antigen-presenting cells like DCs that is required for efficient T-cell stimulation. Mature DCs are then able to initiate the adaptive immune response by activating antigen-specific naïve T-cells to proliferate and produce type II IFN.66,67 The type II IFN receptor is called IFNGR. It activates genes containing a gamma-activated sequence (GAS) through JAK1/2 and STAT1 signaling (Figure 3). Type II IFNs stimulate the adaptive immune response and activate macrophages and NK cells.

Interferon pathway. Type I IFN binds to the IFNAR. IFNAR activates the JAK family members JAK1 and Tyk-2, with subsequent phosphorylation of signal transducer and activator of transcription 1 (STAT1) and 2 (STAT2) proteins. These proteins form the complex called ISGF3 when they bind to IRF9 (p48). ISGF3 initiates transcription of several ISGs by binding to ISREs in their promoter regions. The receptor of type II IFN is called IFNGR and also initiates induction of JAK1 and JAK2 recruitment with STAT1 homodimers that activate genes containing a GAS.
The expression levels of ISGs are predictive of the response to immune checkpoint inhibitors in melanoma patients treated with anti-CTLA-4 antibodies. 68 Likewise, tumor samples from melanoma, ovarian, lung, breast or colorectal cancer can be classified according to high or low expression levels of IFN-stimulated viral defense genes for example IRF7, RIG1 STAT1, IFNB1, IFI6 induced by the hypomethylating agent (HMA) 5-aza-cytidine (5-aza). 69 Our own work shows that lung cancer and melanoma patients with high tumoral expression of IFN-γ have a better outcome with immunotherapy compared with patients with low IFN-γ expression. 70
The role of DNA methylation in innate immune responses
Epigenetic modulator drugs can restore immunogenicity and immune recognition of tumors. Therefore, there is an increasing interest in combined epigenetic therapy and immunotherapy. 71
DNA methyltransferases (DNMTs) are important players in epigenetic modulation of the innate immune response. In non-small cell lung cancer with mesenchymal phenotype, STAT3 activates DNMT1, which methylates the promoter regions of RIG1 as well as IRF1, immunoproteasomes (PSMB8, PSMB9) and HLA molecules leading to the reduction of their expression. 72 STAT3 also inhibits STAT1 expression, a key regulator of the antigen-presentation machinery in epithelial cells.70,72
HMAs such as the nucleoside analogs of cytidine,5-aza and 5-aza-2-deoxycytidine (5- aza-2dc or decitabine) inhibit DNMTs and are currently approved for the treatment of hematologic malignancies.73,74 HMAs activate the innate immune response through PRRs, but they also have several other activities that make them a good partner to combine with immune checkpoint inhibitors. For instance, HMAs reactivate silenced tumor suppressor genes that encode proteins which limit the proliferative and survival capacity of a cell.75,76 HMAs induce T-cell responses by stimulating HLA I expression. For example, in brain tumors, decitabine promotes the surface presentation of tumor-associated peptides in the context of HLA I and is thus a good candidate to be combined with other immunotherapeutic regimen. 77 Furthermore, treatment of tumor cell lines with the DNA methyltransferase inhibitor 5-azacytidine (AZA) enrich tumors in genes involved in immunomodulatory pathways, defining an ‘AZA IMmune gene set (AIMs)’ that can classify primary tumors into ‘high’ and ‘low’ AIM gene expression subsets (see Table 1, in Li and colleagues 78 ). HMAs induce the expression of chemokines that ultimately re-educate tumor cells to become more immunogenic.71,79
Last but not least, HMAs have antitumor activities by causing the re-expression of endogenous retroviruses (ERVs) in preclinical cancer models. Human ERVs (HERVs) are retroviral elements that have been fixed within the human genome through evolution. About 8% of the human genome is composed of these HERVs, most of which are non-functional due to the accumulation of mutations and epigenetic control. Chiappinelli and colleagues showed in preclinical models that HMAs through ERV expression induce viral mimicry and IFN signaling to increase tumor immunogenicity and recognition. 13 Other investigators have reported similar findings in colon cancer, demonstrating an IFN response through activation of MDA5 by dsRNA. 80 This effect, through induction of HERVs, was synergistic with the effect of anti-CTLA-4 or anti-PD-1/PD-L1 antibodies.79,13,81 One way to activate TLRs is through induction of dsRNA from HERVs using low doses of HMAs. HERV-derived viral transcripts then increase within the cytosol leading to RIG1, MDA5 and TLR3 activation and subsequently type I IFN production. 82 The HMA-induced upregulation of HERVs is synergistic with anti-PD-1/PD-L1 antibodies 80 and anti-CTLA-4 antibodies.82,83
In a subset of non-small cell lung cancer patients, 20% of durable responses were observed when the anti-PD-1 antibody was given after tumor progression under low dose of HMAs, indicating that HMAs may prime tumors for a subsequent response to immunotherapy.79,84 Based on the above and other evidences, there is currently a long list of clinical trials in several types of tumors combining immune checkpoint inhibitors with epigenetic drugs (Table 1 and Table 1 in Dunn and colleagues 71 ).
Interestingly, apart from HMAs, cyclin-dependent kinase 4/6 (CDK4/6) inhibitors also suppress DNMT1 and induce viral mimicry. The combination of immune checkpoint inhibitors with CDK4/6 inhibitors was found to be synergistic in vitro and in vivo85,86 and clinical trials are now ongoing with this combination [ClinicalTrials.gov identifiers: NCT02791334, NCT02079636, and NCT02779751].
Other factors in the innate immune response and antitumor therapy
NK cells play an important role against tumor cells. Cells infected by viruses, as well as cancer cells, may downregulate surface MHC class I molecules in order to avoid recognition by CD8+ T-cells. 87 Paradoxically, this phenomenon makes them susceptible to NK cells, that are activated when their membrane receptors, killer-cell immunoglobulin-like receptors (KIRs), are not bound to MHC class I molecules. Some cancer cells bypass NK control because they downregulate specific MHC class I molecules, that are needed to present peptides to CD8+ T-cells, while they still express MHC class I peptides that serve as KIR ligands. 88
Viral restriction factors comprise a group of several proteins expressed by cells to suppress viral replication. These restriction factors constitute an early defense against viral infections, and are partly induced by IFNs. 89 The best characterized restriction factors are: apolipoprotein B messenger RNA editing enzyme catalytic polypeptide-like3 (APOBEC3) proteins, tripartite-motif-containing 5a (TRIM5a), SAM domain and HD domain-containing protein 1 (SAMHD1), Schlafen 11 (SLFN11) and Tetherin. 89 Their role in the immune response to cancer cells has not been studied, however it is known that some of them are involved in the process of somatic mutagenesis during tumor development90,91 APOBEC3 alterations (mutations or overexpression) in cancer cells have been linked to a specific hypermutation status named ‘kataegis’ that correlates with responsiveness to immunotherapy. 92 Finally, a strong correlation has been reported among APOBEC3 proteins, IFNγ and PD-L1 expression in cancer cells. 92
There are several compounds in development with the aim to activate an innate immune response and direct CD8+ T-cells into tumors (Table 1). Such strategies include the combination of anti-PD-1 antibodies with oncolytic virotherapy which has shown impressive results in early clinical trials. 93 Melanoma patients treated with the combination of the anti-PD-1 antibody pembrolizumab plus talimogene laherparepvec (T-VEC) had an objective response rate of 62% with 32% of complete responses. 10 Although the exact mechanism by which oncolytic viruses increase the activity of immune checkpoint blockade is unknown, biopsies taken from patients before treatment and after 6 weeks, demonstrated an increase in CD8+ T-cell infiltration as well as increased expression of IFN-γ suggesting a role of innate immune response activation by T-VEC. 10 Similarly, a hybrid of an oncolytic, nonpathogenic poliovirus (PV) and a human rhinovirus (PVSRIPO) demonstrated an increased release of DAMPs through tumor lysis activating a type I IFN response in DCs in different cancer models. 9
Conclusions and future perspectives
Innate immune responses in the form of IFN production and their subsequent effects provide the appropriate microenvironment for the efficient stimulation of adaptive responses that are key in fighting microbial infections. As tumors are derived from tissue cells that carry mainly self-antigens, they are expected to be purely immunogenic, and may lack innate immune activation, at least initially. Subsequently, the growing tumor can exploit all of the immune suppressive mechanisms that are well described in chronic infections.
Recent clinical trials and preclinical cancer models now impressively demonstrate that stimuli of innate immunity in combination with other immunotherapeutic regimens can significantly augment tumor-specific responses that translate into increased response and cure rates (Figure 1). Thus, suppressive mechanisms and low immunogenicity may be overcome at least when sufficient numbers of tumor neoantigens are present in the tissue.
While presently the PRRs agonists and HMAs stand out, other drugs and drug combinations are under study in numerous trials (Table 1). Furthermore, there are now ongoing trials that include cancer patients with persistent virus infections that have previously been excluded. For example, we are currently exploring several above-mentioned factors and their relationship with innate immune responses in a phase II Spanish Lung Cancer Group clinical trial. The anti-PD-L1 inhibitor durvalumab is given to HIV-1-infected patients with solid tumors [ClinicalTrials.gov identifier: NCT03094286]. The trial enables us to investigate the effect of immune checkpoint inhibition in reversing cancer pathways and to characterize HIV-specific T-cell functions during persistent HIV infection. The study results of this trial and of others are eagerly awaited.
It seems that after many years of stagnation, we finally face most fruitful and exciting times in tumor immunology.
Footnotes
Funding
AM is supported by a grant from the Spanish Ministry of Economy, Industry and Compet-itiveness and FEDER grant no. SAF2016-75505-R (AEI/MINEICO/FEDER, UE), and the ‘María de Maeztu’ Programme for Units of Excellence in R&D (MDM-2014-0370).
Work in Dr Rosell’s laboratory is partially supported by a grant from La Caixa Foundation, and by a European Grant (ELBA no 765492).
Conflict of interest statement
The authors declare that there is no conflict of interest.