
Abstract
Aberrations in the homologous recombination repair (HRR) pathway in prostate cancer (PCa) provide a unique opportunity to develop therapeutic strategies that take advantage of the reduced tumor ability to repair DNA damage. Poly-ADP-ribose polymerase (PARP) inhibitors (PARPi) have been shown to prolong the survival of PCa patients with HRR defects, particularly in those with Breast Cancer type 1 susceptibility protein/Breast Cancer type 2 susceptibility protein alterations. To expand the benefit of PARPi to patients without detectable HRR alterations, multiple preclinical and clinical studies are addressing potential synergies between PARPi and androgen receptor signaling inhibitors, and these strategies are also being evaluated in combination with other drugs such as immune checkpoint inhibitors. However, the effectiveness of these combining therapies could be hindered by multiple mechanisms of resistance, including also the role played by the immunosuppressive tumor microenvironment. In this review, we summarize the use of PARPi in PCa and the potential synergies with different molecular pathways. However, numerous unanswered questions remain, including the identification of the patient population that could benefit most from PARPi, determining whether to use PARPi as monotherapy or in combination, and finding the optimal timing of PARPi, expanding the use of genomic tests, and optimizing combination therapies.
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer and the second leading cause of cancer-related mortality in men worldwide. 1 Patients with hormone-sensitive prostate cancer (HSPC) usually respond to androgen deprivation therapy (ADT), but eventually acquire resistance and progress to lethal stage of castration-resistant prostate cancer (CRPC) with (mCRPC) or without detectable metastases (nmCRPC) on conventional imaging. 2 Over the years, several clinical and molecular studies have provided new insights into the landscape of PCa staging to better subclassify the prostate disease for therapeutic interventions. Despite considerable advances in the treatment options for prostate tumor, including the integration of radiopharmaceuticals3,4 and targeted therapies, such as poly-ADP-ribose polymerase (PARP) inhibitors (PARPi),5,6 an improved understanding of the mechanisms of disease progression and therapy resistance is leading to develop novel treatment strategies, thus helping our progress toward precision medicine. In this context, we review the interplay among androgen receptor (AR) signaling, PARP pathway, and the tumor microenvironment (TME) in PCa with the aim of highlighting adaptive resistance mechanisms that can play an important role in primary or acquired therapeutic resistance. We also discuss novel strategic combinations of androgen receptor signaling inhibitor (ARSI), PARPi, and immune checkpoint inhibitors (ICIs) to prevent and overcome resistance and ameliorate sensitivity to targeted therapies in PCa.
AR and its inhibitors in PCa
Pathophysiology of AR in PCa
AR-driven signaling pathway plays a pivotal role in both the physiology and pathophysiology of the prostate gland and other male reproductive organs. It is mainly involved in normal processes of cell differentiation and growth, as well as in cancer initiation and progression where AR signaling is active and supports the PCa cells survival.7,8
AR is a 110 kDa protein (919 amino acids, aa) encoded by the AR gene located on the X chromosome (Xq12), over 90 kb in length, and consists of 8 exons and 7 introns. It is a ligand-activated nuclear transcription factor belonging to the steroid hormone receptor family. Similar to other steroid receptors, AR is characterized by four distinct functional regions: the N-terminal transactivation domain (NTD); the DNA-binding domain (DBD); the hinge region; and the ligand-binding domain (LBD).7,9,10
The NTD (encoded by exon 1, 1–537 aa) is generally constitutively active and implicated in cellular transcription complexes. It harbors transcriptional activation function-1 (AF-1) with its two major transactivation units (TAU): TAU-1 (142–485 aa) and TAU-5 (351–528 aa). TAU-5 drives AR-dependent transcriptional activation and it has been associated with aberrant AR activation that drives resistance to ADT in CRPC cells.7,11 The DBD (encoded by exons 2 and 3, 538–624 aa) allows AR-DNA binding. It contains two zinc finger motifs required for interaction with specific DNA sequences and facilitates receptor homodimerization. The hinge region (encoded by exon 3–4, 625–669 aa) separates the DBD from the LBD and encodes the nuclear translocation signal required for AR nuclear import. The LBD (encoded by exons 5–8, 626–919 aa) contains transcriptional activation function-2 (AF-2) and allows the binding of androgen ligands [Figure 1(a)].7,12–14

AR gene, protein, and signaling pathway. (a) The AR gene is located on the X chromosome (Xq12) and consists of eight exons. AR protein contains four functional regions: the NTD (encoded by exon 1) that includes the transcriptional AF-1 and two TAUs: TAU-1 and TAU-5; the DBD (encoded by exons 2–3), the hinge region (encoded by exons 3–4), and the LBD (encoded by exons 4–8) that contains the transcriptional AF-2. (b) In the cytoplasm, in its inactive form, AR is sequestered by the complex of the chaperone HSPs. Upon binding the DHT, a conformational change in the AR occurs. The receptor dissociates from the HSPs, translocate into the nucleus, and dimerizes. AR homodimer binds to ARE sited in the promoter regions of AR target genes and promotes their transcription. These genes (i.e. KLK3/PSA, TMPRSS2) are mainly involved in cell proliferation and survival. AR signaling can be inhibit by ARSI agents, such as: Abiraterone, which targets the adrenal androgen-mediated signaling axis by inhibiting the androgen synthesis, and Enzalutamide, Apalutamide, and Darolutamide which impair the translocation of AR into the nucleus.
AR responds to androgenic steroid hormones, such as testosterone and 5α-dihydrotestosterone (DHT). AR is in the cytoplasm in an inactive form, sequestered by the complex of the chaperone heat shock proteins (HSPs), including the HSP90. In presence of DHT, the binding of the androgen to the LBD of AR results in a conformational change that leads to dissociation of the receptor from the chaperone protein complex and its translocation into the nucleus where it dimerizes with a second AR protein. AR homodimers bind to androgen response elements in the promoter regions and regulates the transcription of AR target genes, such as KLK3, which encodes prostate specific antigen (PSA) and transmembrane protease serine 2 (TMPRSS2) and genes involved in the DNA damage response (DDR). Transcriptional regulation of these target genes contributes to the differentiation and growth of the prostatic epithelial cells [Figure 1(b)]. 12
Therefore, the AR signaling axis plays a critical role in the development and progression of PCa and represents a target for anticancer therapies. 9 Although a majority of patients initially respond to ADT, most will eventually develop castrate resistance. Over the years, several findings have shown that AR signaling persists during castration, mainly due to the emergence of different mechanisms of therapeutic resistance, such as constitutively activation of AR splice variants, and the presence of AR gene amplification and/or point somatic mutations.15–17
ARSI: Clinical study
The ADT still now represents the cornerstone for PCa treatment. Reducing testosterone, ADT deprives PCa cells of the hormone-dependent signals, essential for tumor survival and growth. Several mechanisms have been proposed for driving the onset of castration resistance, including AR gene amplification and protein overexpression, gain-of-function mutations, AR-variant development, co-activator overexpression, and ligand-independent AR transactivation. Therefore, castration resistance needs agents capable of overcoming these resistance mechanisms. At the same time, some disease settings, such as metastatic hormone-sensitive prostate cancer (mHSPC), require therapeutic intensification approaches with the addition of other agents to potentiate the ADT activity, such as ARSIs, starting from the diagnosis. 18
So far, four ARSI approved by Food and Drug Administration (FDA)/European Medical Agency (EMA) have been developed in different settings of PCa treatment (Table 1):
- Abiraterone acetate targets the adrenal androgen-mediated signaling axis, acting as pregnenolone analog and blocking cytochrome P450 (CYP17A1), the rate-limiting enzyme for the adrenal precursors for testosterone and DHT synthesis. It is used in mHSPC and mCRPC.
- Enzalutamide is a second-generation ARSI that blocks several steps in the AR signaling pathway, including androgen binding to AR, nuclear translocation of activated AR, and binding of activated AR with DNA. It is indicated in mHSPC, nmCRPC, and mCRPC.
- Apalutamide is a non-steroidal second-generation ARSI that directly inhibits the ligand-binding domain of AR, allowing the inhibition of its nuclear translocation, DNA binding, and AR-mediated transcription. Apalutamide is used in the mHSPC and nmCRPC settings.
- Darolutamide is a non-steroidal ARSI acting through the inhibition of testosterone-induced AR translocation to the nucleus, thus decreasing the AR-dependent gene expression. It is used in patients with mHSPC or nmCRPC.
Approved ARSI by FDA/EMA and corresponding registration trials in different PCa settings.
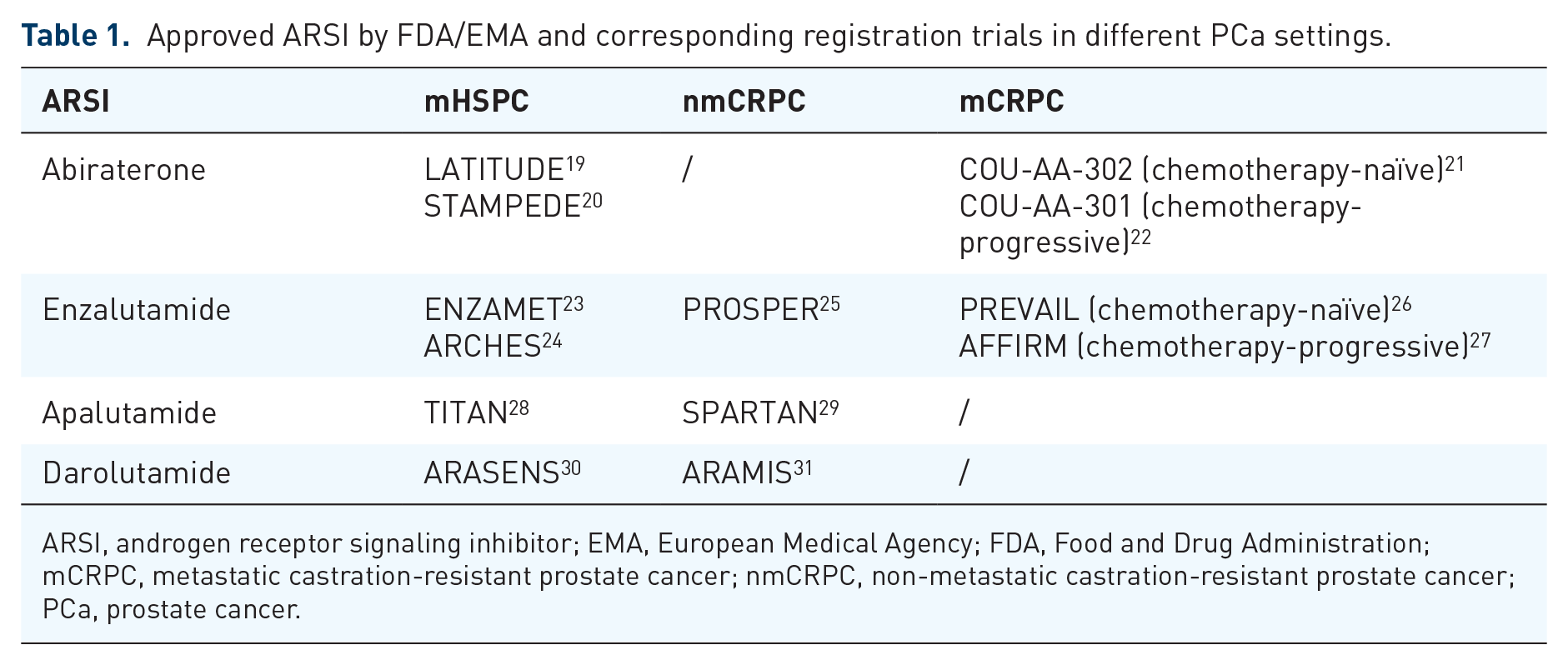
ARSI, androgen receptor signaling inhibitor; EMA, European Medical Agency; FDA, Food and Drug Administration; mCRPC, metastatic castration-resistant prostate cancer; nmCRPC, non-metastatic castration-resistant prostate cancer; PCa, prostate cancer.
The role of AR and the interplay with DNA damage repair in PCa
Genomic instability is a hallmark of most malignancies. PCa shows a marked mutational burden with a high rate of genomic instability, including large numbers of deletions, insertions or duplications, amplifications, and chromosomal rearrangements.32,33 Genomic profiling studies using whole exome sequencing and whole genome sequencing have identified the presence of a high frequency of genetic aberrations in PCa, particularly in its more aggressive form, the mCRPC. These aberrations mainly involve the AR, TP53, RB1, PTEN, c-MYC, SPOP, and DNA repair genes.34,35
The onset of genomic alterations is related to DNA damage, which can be endogenous and arise from cellular metabolism (e.g. reactive oxygen species and hydrolytic reactions), or exogenous (e.g. chemicals and radiation). DNA lesions impair the replication and transcription machinery, leading to cell cycle arrest or DNA lysis and collapse and the accumulation of genomic alterations drives the carcinogenesis process.32,33
The role of PARP1 in DDR
In order to preserve cells from potentially cancerous mutations, the DDR signaling pathway has a crucial role. DDR monitors DNA integrity: in the presence of DNA damage, it induces cell cycle arrest, the recruitment of repair factors to the site of the lesion and promotes its repair. 36
In eukaryotic cells, the most common DNA damages are the single-strand breaks (SSBs) and the double-strand breaks (DSBs), respectively, repaired through different mechanisms. SSBs are repaired by base excision repair (BER), nucleotide excision repair, and DNA mismatch repair pathways. A pivotal role in this process is played by PARP, an ADP-ribosyl transferase belonging to the PARP family which is composed of 17 proteins, including PARP1 and PARP2, two DNA damage sensors. 37
PARP1 is a nuclear-localized enzyme involved in the regulation and preservation of chromatin structure promoting genomic integrity. 38 PARP1 recognizes the SSB site, binds to DNA through its DNA-binding domain, cleaves its cofactor nicotinamide adenine dinucleotide (NAD+), and catalyzes poly (ADP-ribose)-ylation (PARylation) by transferring ADP-ribose to itself or to other target proteins with the formation of a covalent bond between the protein and ADP-ribose or subsequent ADP-ribose polymers. The PARylation reaction acts as a signal to recruit other factors involved in DNA repair to the site of damage, such as DNA ligase III, DNA polymerase beta, and scaffold proteins (e.g. XRCC1), so facilitating the assembly and the activation of the BER machinery.38,39
Failure of SSB repair systems leads to the accumulation of SSBs that can turn into DSBs during DNA replication. DSBs are repaired via two main pathways: the homologous recombination (HR) and the non-homologous end joining (NHEJ) systems.32,37,40 HR is activated during S and G2 phases of the cell cycle and requires a DNA template for error-free repair.33,41 The main actors of the DSBs repair machinery are serine–threonine protein kinases belonging to the phosphatidylinositol 3-kinase-like protein kinase family, which include Ataxia Telangiectasia Mutated (ATM), Ataxia Telangiectasia and Rad3-related protein (ATR), and DNA-dependent protein kinase (DNA-PK).36,42
It is well known that PARP1 is also involved in DSBs repair systems. PARP1 detects, binds DNA damage sites, and promotes the recruitment of the MRE11–RAD50–NBS1 (MRN) complex. In turn, the MRN complex recruits and activates the protein kinase ATM. In addition, PARP1 is involved in recruiting Breast Cancer type 1 susceptibility protein (BRCA1), Partner and Localizer of BRCA2 (PALP2), and Breast Cancer type 2 susceptibility protein (BRCA2) favoring the assembly of the BRCA1/PALB2/BRCA2 complex. BRCA2 promotes RAD51 loading at the damaged site to initiate invasion and strand repair through D-loop formation and DNA synthesis (Figure 2).32,37,43,44 Moreover, a key role is also played by Checkpoint kinase 1 (Chk1) and Checkpoint kinase 2 (Chk2), two serine–threonine kinases that arrest the cell cycle, thus allowing time for repair of DNA damages. In addition, Chk2 also interacts with p53 and p53 stabilization by Chk2 leads to cell cycle arrest in the G1 phase.33,36

DSBs repaired by HR. When a DSB occurs, PARP detects the breaks and binds DNA damage site by promoting the recruitment of the MRN complex. In turn, the MRN complex recruits and activates the protein kinase ATM. PARP1 also enrols BRCA1, PALP2, and BRCA2 favoring the assembly of the BRCA1/PALB2/BRCA2 complex. Thus, BRCA2 promotes RAD51 loading at the damaged site to initiate invasion of the intact sister chromatid and repairs the strand through D-loop formation and DNA synthesis.
Mutations of DDR genes and PARPi
Next generation sequencing (NGS) analyses have shown that approximately 10% of primary tumors and 25% of PCa metastases have alterations in genes involved in DDR. These genomic aberrations involve several genes including BRCA1/2, ATM, CHEK2, PALB2, RAD50, RAD51C; among these, BRCA2 mutations are the most frequent in PCa. 45
In 2005, two crucial studies demonstrated that the onset of BRCA1 or BRCA2 mutations sensitized cells to PARPi, causing chromosomal instability, cell cycle arrest, and apoptosis.46,47 PARPi leads to the accumulation of single-strand DNA breaks that degenerate during DNA replication into DSBs. In homologous recombination repair (HRR) competent cells, DSBs are physiologically repaired by the HR pathway. However, in homologous recombination deficient cells with BRCA1/2 mutations PARPi prevents repair of tumor DNA and leads to ‘Synthetic Lethality’ cell death. The concept of synthetic lethality occurs when two non-lethal defects combine together and give rise to a lethal phenotype (Figure 3).36,48
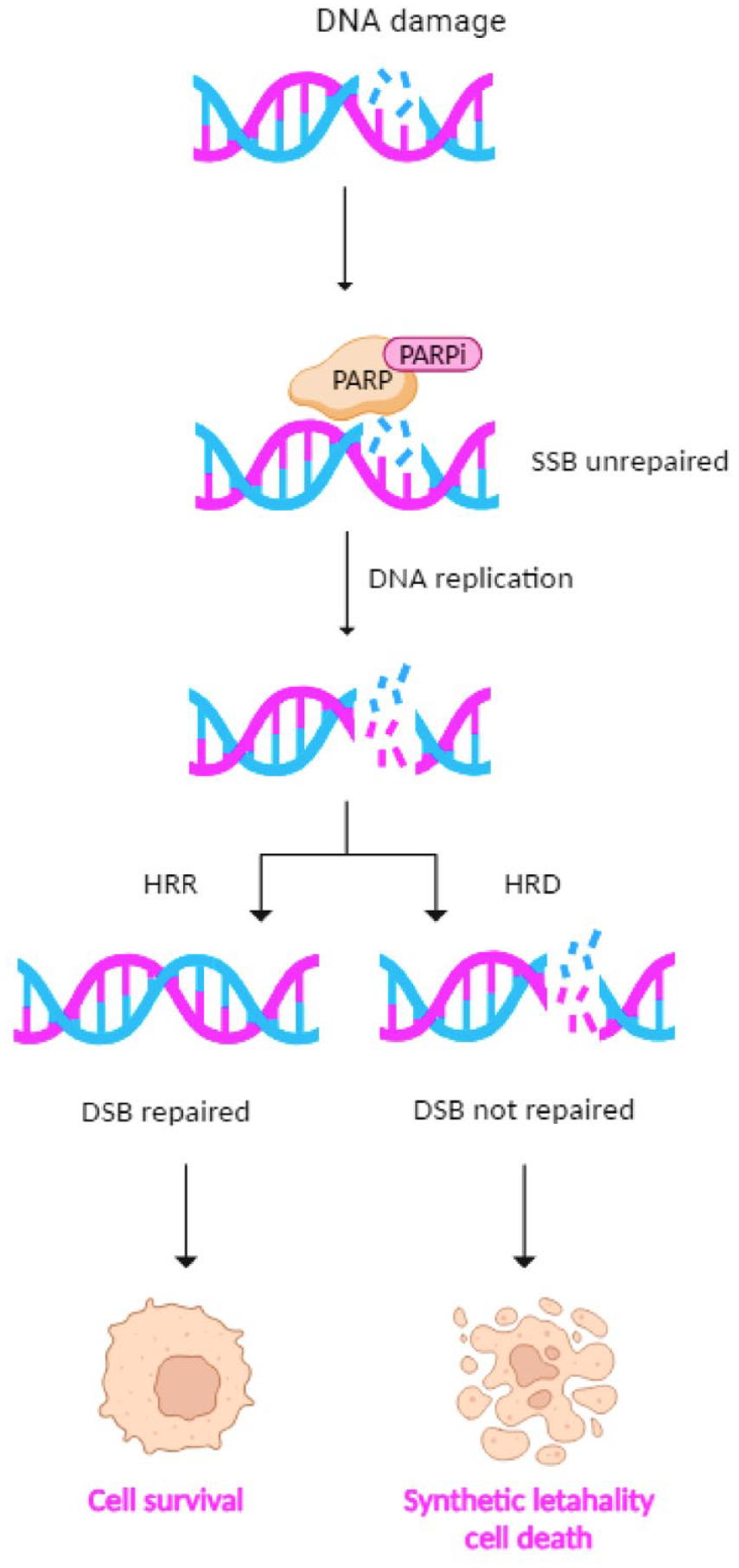
PARPi effects in HRR competent and HRD cells. In HRR-competent cells, PARPi prevents DNA SSBs repair. SSBs during DNA replication degenerate into DNA DSBs and cells repair the DNA damage using HRR. In HRD cells in the presence of PARPi, neither SSB nor DSB could be repaired. This leads to cell death through synthetic lethality.
The interplay between AR and DDR
The role of AR in DDR is not yet fully elucidated even though several studies have suggested a mutual interaction between activation of AR signaling the regulation of DDR gene expression. Polkinghorn et al. have demonstrated that AR regulates upstream transcription of several genes involved in DDR pathway by defining an ‘AR-associated DNA repair gene’ signature of 144 DDR genes (e.g. RAD51, POLE2, CHEK1, PARP1) significantly associated with canonical AR activation. Moreover, they found that AR signaling increases the expression of DDR genes promoting tumor radioresistance by accelerating the repair of radiotherapy-induced DNA damage. This evidence shows a potential synergy between ADT and radiation therapy, suggesting a potential benefit of ADT treatment in AR-positive patients undergoing radiotherapy. 49
Goodwin et al. have found that AR activity is induced to DNA damage and in turn AR activation promotes higher expression of genes involved in the DDR program. The AR activity and the expression of the main clinically relevant AR target genes, such as KLK3/PSA, TMPRSS2, and FKBP5, has been investigated in CRPC cells after ionizing radiation (IR) exposure. Both TMPRSS2 and FKBP5 transcripts increased after IR, while KLK3/PSA levels remained generally unchanged. In order to evaluate the specificity of this response, Goodwin and colleagues evaluated the impact of doxorubicin, a chemotherapy agent known to cause DNA DSBs, on the expression of DDR genes induced by AR. Doxorubicin showed a marked induction of AR activity, similar to that resulting after IR exposure. Furthermore, it is interesting to note that UV treatment, which causes SSBs, did not alter AR target gene expression, demonstrating that AR activity is selectively induced in response to DNA DSBs. As counter evidence, it was further observed that androgen deprivation abrogated the DNA damage-induced upregulation of AR targeted gene expression. 50
As demonstrated by Polkinghorn et al. 49 studies, Goodwin et al. have also demonstrated that the activation of AR signaling induces the expression of three main genes involved in DDR: PRKDC gene, which encodes for the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), a protein required for NHEJ repair of DNA DSBs; XRCC2 and XRCC3 genes encoding for the protein of RAD51 family that participates in HR to maintain chromosome stability and repair DNA damages.42,50,51 In addition, accumulated DNAPKc enhances AR activity, creating a positive feedback circuit through which androgens promote DNA repair. 50
Overall, these findings identify the AR signaling as a key regulator of the DDR system, thus promoting tumor cell survival and likely inducing resistance to cytotoxic agents.
Interaction PARPi of ARSI: Pharmaceutical induction of ‘BRCAness’
The interplay between PARP1 and AR
It is well-established that PARP-1 is involved in the DDR pathway, but its impact on transcriptional regulation is not yet well defined. Schiewer et al. 52 have demonstrated that PARP1 supports tumor growth and progression through AR transcriptional regulatory functions in PCa cell models. They have evaluated the impact of PARP1 on the response to DNA damage induced by IR and docetaxel treatment. Both HSPC and CRPC cells have been treated with a clinically relevant PARPi, ABT888. Exposure to ABT888 as a single agent reduced the growth of AR-positive HSPC and CRPC cells and sensitized both PCa cell models to IR and Docetaxel. In contrast, no effect was observed in AR-negative PCa cells. This evidence suggests that PARP inhibition cooperates with genotoxic agents affecting the cell survival of PCa cells expressing AR.52,53
Investigating the effect of PARP1 on AR function, they have observed that ABT888 significantly reduced the expression of AR target genes mainly involved in PCa progression, such as, KLK3/PSA, TMPRSS2, and FKBP5, without altering AR protein levels. Using chromatin tethering assays and chormatin immunoprecipitation (ChIP) assay, they further demonstrated that PARP influences AR activity by promoting an open chromatin structure that facilitates AR transcriptional activity in both HSPC and CRPC cells.52,54 In addition, a recent study has demonstrated that PARP1/2 are also involved in regulating the transcriptional activity of variant forms of AR and that both androgen target genes and DDR genes show attenuated expression following enzymatic blockade with PARPi. 55
Furthermore, Schiewer et al. also revealed that in CRPC cell lines PARP1 is highly PARylated, compared to HSPC cells, thus suggesting that PARP1 activity is upregulated in castration resistance models and ligand-independent AR function along with chromatin occupancy depends on PARP1 enzymatic activity. Interestingly, in in vivo studies in patient-derived xenografts mouse models have corroborated in vitro experimental data showing that the suppression of PARP1 induces a reduction in AR-dependent target gene expression, AR function, and AR-driven tumor progression. Finally, immunohistochemical assays in the ex vivo patient-derived primary PCa models treated with ABT888, to suppress the PARP activity, showed a significant reduction of PARP global level, a marked decrease of the proliferation marker Ki67 and lower expression of AR target genes. 52
Taken together, this evidence identifies novel functions of PARP1 in promoting tumor progression through AR-dependent transcriptional regulatory functions and elect PARP1 as a potential therapeutic target to inhibit PCa progression in cooperation with ARSI.
Experimental evidence of pharmaceutical induction of ‘BRCAness’
Several lines of evidence suggest that AR targeting may represent a strategy to affect DDR pathways. Experimental data obtained by Li et al. showed the effect of Enzalutamide, a second-generation ARSI, on AR-regulated HR genes in different PCa cell lines with variable androgen sensibility. They investigated the expression of five HR genes including BRCA1, RAD51AP1, RAD51C, RAD54L, and RMI2. The treatment with Enzalutamide downregulated this specific set of HR genes in AR-positive androgen-dependent cell lines, but the Enzalutamide was ineffective in androgen-independent PCa cell lines. Unexpectedly, treatment with PARPi, such as Olaparib, suppressed the expression of these HR genes in both androgen-dependent and -independent cell lines. Furthermore, combined treatment further downregulated these HR genes in an androgen-sensitive model. Moreover, the analysis of DNA damage biomarkers, such as γH2AX and RAD51, showed how the combination treatment with ARSI and PARPi in HSPC cells significantly increased DNA damage by inducing apoptosis. Together, this evidence suggests that the cytotoxic effects of the ARSI/PARPi combination are mediated by their synergic activity inducing HR inefficiency also called ‘BRCAness’. 56
A contribution to the understanding of the pharmacological induction of BRCAness was also given by a recent study by Dong et al. Their findings support the therapeutic efficacy of combined treatment with Enzalutamide and Olaparib in AR-positive PCa cells in terms of reduction of cell viability, increase in γH2AX expression and induction of apoptosis by affecting NHEJ and up-regulating pro-apoptotic genes and down-regulating anti-apoptotic genes. 57
The synergism of the Enzalutamide and Olaparib treatment was also tested by Li and colleagues in in vivo using both a patient-derived subcutaneous xenograft tumor model and two orthotopic PCa cell line xenograft models (AR-positive, androgen-dependent and -independent). In the subcutaneous xenograft tumor model and orthotopic PCa cell line xenograft hormone-sensitive models, the treatment with the single agents was sufficient to suppress tumor growth. The effect was enhanced in the presence of the combined treatment, especially with an Enzalutamide pretreatment, showing a reduction of the levels of the proliferation marker Ki67. On the contrary, in the orthotopic PCa cell line xenograft hormone-independent model, PARPi showed similar efficacy to the combined treatment. 56
Overall, pharmaceutically (ARSI)-induced ‘BRCAness’ may sensitize PCa to PARP inhibition, expanding the clinical use of PARPi even in patients who do not carry BRCA1/2 mutations.
Clinical studies of ARSI + PARPi
Used as single agents, PARPi increased survival and response rates in mCRPC patients. 58 So far, two PARPi have been authorized by the FDA and the EMA: Olaparib, approved by FDA for patients with HRR alterations after ARSI progression, and by EMA with a restriction to BRCA1/2-mutated patients; Rucaparib, FDA-approved for BRCA1/2-mutated patients progressing to ARSI and taxanes. After being tested as single agents, PARPi started to be evaluated in combination with agents having different mechanisms of action in phase II or III randomized clinical trials, and three combinations of PARPi and ARSI were approved: Olaparib plus Abiraterone, Niraparib plus Abiraterone, and most recently Talazoparib plus Enzalutamide (only FDA-authorized) (Table 2).
Studies of PARPi plus ARSI combination in mPC.

AEs, adverse events; ALT, alanine transaminase; ARSI, androgen receptor signaling inhibitor; ATM, Ataxia-Telangiectasia Mutated; BRCAm, BRCA mutant; CI, confidence interval; CRR, complete response rate; HR, hazard ratio; HRR, homologous recombination repair; ITT, intention to treat; mCRPC, metastatic castration-resistant prostate cancer; mHSPC, metastatic hormone-sensitive prostate cancer; mOS, median overall survival; mPC, metastatic prostate cancer; mPFS, median progression-free survival; NR, not reached; ORR, overall response rate; PARPi, poly (ADP-ribose) polymerase (PARP)-inhibitors; PBO, placebo; PSA-RR, PSA-response rate; QoL, quality of life; rPFS, radiographic progression-free survival; TRAE, treatment-related adverse event.
The MAGNITUDE (NCT03748641) study randomized 670 naïve mCRPC patients to Abiraterone 1000 mg once daily (OD) (plus Prednisone 5 mg bis in die - BID) and Niraparib 200 mg OD or placebo (PBO). Abiraterone could have been started 4 months before the inclusion in the study. Patients were selected for HRR status. The radiographic progression-free survival (rPFS) was the primary endpoint, with a hierarchical analysis first in the BRCA1/2-mutated subgroup, then in the other HRR-mutated patients; overall survival (OS), time-to-chemotherapy, and time-to-symptomatic progression were the secondary endpoints. The HRR-negative cohort was stopped for futility after the pre-planned interim analysis. After a median follow up (mFU) of 18.6 months, Niraparib plus Abiraterone significantly prolonged rPFS compared to Abiraterone plus PBO [median rPFS 16.5 versus 13.7 months; hazard ratio (HR), 0.53; 95% confidence interval (CI), 0.36–0.79; p = 0.0014] in the HRR-positive cohort. In the BRCA1/2-mutated subgroup, median rPFS was significantly longer in the Abiraterone plus Niraparib than in the Abiraterone plus PBO group (16.6 versus 10.9 months; HR, 0.53; 95% CI, 0.36–0.79; p = 0.001). Also, the other secondary endpoints were significantly improved with Abiraterone plus Niraparib compared to Abiraterone plus PBO.
A higher ORR was observed in the experimental group (52% versus 31% in BRCA mutated patients and 60% versus 28% in HRR-positive patients), with 18% versus 14% in BRCA mutated patients and 22% versus 11% in HRR-positive patients with a complete response. AEs were reported in 99.1% versus 94.1% of patients, with ⩾G3 AEs in 67.0% (commonly, anemia) versus 46.4% (commonly, hypertension). The cases of dose reduction were approximately 19.8% versus 3.3% of cases and treatment interruption in 10.8% versus 4.7%. Anyway, quality of life (QoL) questionnaires showed comparable results between the two cohorts. 59 Nineteen patients died in each group: in the Abiraterone plus Niraparib cohort, infections were the most frequent causes of death, in the Abiraterone plus PBO group, cardiac disorders were the main death reasons. In the second interim analysis, Niraparib plus Abiraterone prolonged rPFS to 19.5 months in the BRCA-mutated subgroup versus 10.9 (HR, 0.55; 95% CI, 0.39–0.78; p = 0.0007).60,61 At the ESMO 2023, the OS analysis at a mFU of 35.9 months of BRCA-mutated patients evidenced a mOS of 30.4 versus 28.6 months (HR, 0.79; 95% CI, 0.55–1.12; p = 0.18). 61
In the PROpel (NCT03732820) trial, mCRPC patients were eligible if they had not previously received Abiraterone or had stopped another ARSI at least 12 months before enrolment. Docetaxel was allowed in the hormone-sensitive setting. In total, 796 patients were randomized to Abiraterone (1000 mg OD) plus Olaparib 300 mg BID (n = 399) versus PBO (n = 397). Patients were not selected for HRR status, but around one out of three had HRR mutations, and 10% were BRCA-mutated. Adding Olaparib to Abiraterone prolonged rPFS (the primary endpoint) compared to PBO, with a median rPFS of 24.8 versus 16.6 months (HR, 0.66; 95% CI, 0.54–0.81; p < 0.0001). The benefit was consistent among all subgroups, although it was more significant in patients with BRCA mutations (NR versus 8.4 months; HR, 0.23; 95% CI, 0.12–0.43), or carrying HRR alterations (NR versus 13.9 months; HR, 0.50; 95% CI, 0.34–0.73) than in those without genetic alterations (24.0 versus 19.0 months; HR, 0.76; 95% CI, 0.60–0.97).62,63 After 36.6 months of mFU, mOS (secondary endpoint) was longer in the Abiraterone plus Olaparib group (42.1 months) than PBO plus Olaparib (34.7 months; p = 0.054), with medians NR in the HRR-positive and BRCA-mutated subgroups. 64 More AEs were reported in the Abiraterone plus Olaparib (47%) than in the Abiraterone plus PBO group (38%), and a higher discontinuation rate emerged in the former group (13.8% versus 7.8%). QoL results were comparable between the two cohorts.64–66
The TALAPRO-2 (NCT03395197) trial included mCRPC patients not selected for HRR status, randomized to Enzalutamide plus Talazoparib (n = 402) or Enzalutamide plus PBO (n = 403). The primary endpoint was rPFS in patients with evaluable soft tissue or bone lesions. After a mFU of 25 months, median rPFS was NR in patients treated with Talazoparib plus Enzalutamide versus 21.9 months in the PBO plus Enzalutamide group (HR, 0.63; 95% CI, 0.51–0.78; p < 0.001). An increased benefit was observed in HRR mutated patients (rPFS 27.9 versus 16.4 months; HR, 0.46; 95% CI, 0.30–0.70; p < 0.001). The combination treatment achieved a higher ORR than single-agent Enzalutamide plus PBO (61.7% versus 43.9%). AEs ⩾G3 were observed in 71.9% versus 40.6% of patients, including anemia, hypertension, and fatigue. Discontinuation rates did not change significantly between the two groups; however, the median time to QoL deterioration was significantly higher in the combined treatment group compared to the PBO plus Enzalutamide group (38.0 versus 25.0 months, HR, 0.78; 95% CI, 0.62 to −0.99; p = 0.04).67,68
In the BRCAAway (NCT03012321) study, 165 eligible men were registered and underwent NGS/germline testing; 61 patients with homologous recombination repair gene mutation (HRRm) were randomized 1:1:1 to Abiraterone or Olaparib or Abiraterone plus Olaparib. mPFS – the primary endpoint – was 39 months in the combination group, was 14 months in the Olaparib group and 8.4 months in the Abiraterone group. PSA-response rate (PSA-RR) was 95%, 67% and 58% in the Olaparib plus Abiraterone, Olaparib and Abiraterone groups, respectively. Fifty-one patients had treatment-related AEs; most common Grade 3: fatigue n = 3, anemia n = 2 and alanine transaminase (ALT) increases n = 2. 69
Further studies are ongoing including combinations of PARPi with other drugs, or anticipating the associations of PARPi and ARSI in the hormone-sensitive setting63,70–72 (Table 3).
Ongoing trials of PARPi combination in mPC.

Source: Table adapted with permission from Maiorano et al. 73
AKTi, AKT inhibitor; DLT, dose limiting toxicities; mCRPC, metastatic castration-resistant prostate cancer; mHSPC, metastatic hormone-sensitive prostate cancer; mPC, metastatic prostate cancer; MTD, maximum tolerated doses; ORR, overall response rate; OS, overall survival; PARPi, poly-ADP-ribose polymerase inhibitor; PBO, placebo; PK, pharmacokinetics; PFS, progression-free survival; PSA-CR, PSA-complete response; rPFS, radiographic progression-free survival; RP2D, recommended phase II dose.
PARPi in combinations with ICIs and the role of immune microenvironment
Biological rationale for PARPi and ICIs combinations in PCa
Several preclinical studies suggest that PARPi synergize with ICIs through multiple pathways, resulting in reciprocal potentiation of their antitumor activities.74–76 The main mechanisms include: increased DSBs on DNA, programmed death-Ligand 1 (PD-L1) upregulation, and TME modifications [Figure 4(a)–(c)].

The interplay between PARPi and ICIs. (a) After PARPi administration, immune response is activated in many ways: (i) Neo-antigens derived by DSBs accumulate on the cell surface, and are recognized by APCs; (ii) DNA fragments are recognized by cGAS, cGAS activates 2′–5′ cyclic GMP-AMP, cGAMP switches on STING, STING modulates transcription factors (NF-κB, TBK1, IRF3), resulting in the transcription of immunogenic cytokines (IFN, IL-6, TNF-α); (iii) IFN, ATM–ATR–CHEK1, and STAT–IRF increase the expression of PD-L1; (iv) DSBs inactivate GSK3β, responsible for PD-L1 proteasomal degradation, increasing PD-L1 cellular expression. (b) These modifications result in a more immune responsive TME: increased surface neo-antigens, increased PD-L1 expression, cytokines, and chemotactic factors induce an increase in number and function of APCs, T-cells, NK cells, and decrease in immunosuppressive elements such as MDSCs and TAMs M2 type. (c) Targeting PD-L1, PD1, or CTLA4, ICIs unleash the anti-tumor immune response, potentiating the immune activation against tumor cells. Therefore, the combination of these two drug classes could result in a higher anti-tumor immune response.
When DSBs occur after PARPi, DNA fragments accumulate in the cytoplasm. As a result, neo-antigens on the cell surface are recognized by Antigen-presenting cells (APCs) and activate the immune response. Moreover, the stimulator of interferon genes (STING) pathway is activated: DNA fragments are recognized by the cytosolic sensor cGMP-AMP synthase (cGAS), cGAS activates 2′–5′ cyclic GMP-AMP (cGAMP), and cGAMP switches on STING. Then, STING modulates transcription factors such as nuclear factor-kappa B (NF-κB) and interferon regulatory factor 3 (IRF3), resulting in the transcription of related cytokines [interferon (IFN), interleukin (IL)-6, tumor necrosis factor-alpha (TNF-α), chemokines], which promote the immune response by increasing the percentages of tumor-infiltrating T cells.77,78 IFN increases PD-L1 expression on the cell surface, as well. Other studies highlighted the role of these pro-inflammatory cytokines also in controlling angiogenesis, PCa proliferation and transition toward a CRPC phenotype.79,80 In addition, DSBs activate the ATM–ATR–Chk1 sensory system in the nucleus, and STAT/IRF signaling pathway by promoting the PD-L1 expression on the plasma membrane.81,82 Finally, DSBs inactivate glycogen synthase kinase 3-beta, responsible for PD-L1 proteasomal degradation, further increasing PD-L1 cellular expression74,83 [Figure 4(a)]. Therefore, the more DSBs occur, the more PD-L1 is upregulated and expressed. This evidence constitutes a solid rationale for PARPi and ICIs combination.
PARPi impacts on immune cells by reversing the immunosuppressive and immunologically ‘cold’ TME into a more immunoreactive one. PARP inhibition promote the reduction of myeloid-derived suppressor cells (MDSCs) and immunosuppression-related regulatory T cells (T-regs) in tumor tissue and also relieves the shift of tumor-associated macrophages in the anti-inflammatory M2 type, reprogramming macrophages toward an anti-tumor phenotype.84,85 After PARPi administration, CD4+/CD8+ T cells and natural killer cells are recruited at tumor sites, APCs activate effector T cells and release large amounts of pro-inflammatory cytokines, including IL-2, IFNγ, and TNF-α [Figure 4(b)]. 85
Thus, the simultaneous targeting of PD-L1, PD1, or cytotoxic T-lymphocytes associated protein 4 and PARP, may provide a strategy to potentiate the immune activation against tumor cells in PCa, a tumor type with poor response to ICIs as single agents [Figure 4(c)].85,86
Studies of PARPi in combination with ICIs
Several studies have investigated the association of PARPi and ICIs, starting from the mCRPC setting.87,88 In the CheckMate 9KD (NCT03338790) phase II trial, Olaparib plus Nivolumab were administered to mCRPC patients already treated with chemotherapy and ARSI (cohort A1, n = 88), or chemotherapy-naïve and ARSI-pretreated (cohort A2, n = 77). In cohort A1, ORR was 10.3% (ranging from 3.4% in HRR-negative to 17.2% in HRR-positive patients), PSA50-RR was 11.9% (18.2% in HRR-positive versus 5.0% in HRR-negative patients), median rPFS 4.9 months, and mOS 13.9 months. In cohort A2, ORR was 15.4% (range: 5.9–30.5%) with HRR-positive versus HRR-negative 25.0% (8.7–49.1%) versus 5.3% (0.1–26.0%), respectively, range 5.3% in HRR-positive, and a maximum of 5.3% in HRR-negative patients, PSA50-RR was 27.3% (41.9% in HRR-positive, 14.3% in HRR-negative), median rPFS was 8.1 months, and mOS 20.2 months. Nausea, fatigue, and anemia were the most common AEs in the two cohorts. AEs ⩾G3 were observed in almost half of the patients, including anemia, neutropenia, and increased ALT. 89 Although the results of combination treatment were poor in unselected patients, both the response rates and survival outcomes were improved in HRR-positive patients. In the phase Ib/II KEYNOTE-365 trial (NCT02861573), 102 patients (cohort A) with pre-treated mCRPC received Pembrolizumab + Olaparib 400 or 300 mg BID. This cohort represented a heavily pre-treated population, as 92% of patients were previously treated both Docetaxel and Enzalutamide or Abiraterone, 39% Cabazitaxel, and 45% both Abiraterone and Enzalutamide. Study results showed PSA-RR 15%, ORR 8.5%, mPFS 4.5 months, and mOS 14 months. A total of 91% of patients showed AEs, ⩾G3 in 48% of cases. In addition, six drug-related deaths were reported. 90
The combination treatment with PARPi and ICI was further investigated in the phase III KEYLYNK-010 study, which recruited 793 mCRPC patients progressing to ARSI and Docetaxel receiving Pembrolizumab + Olaparib rather than another ARSI. At final rPFS analysis, median rPFS was 4.4 months (95% CI, 4.2–6.0) with Pembrolizumab + Olaparib and 4.2 months (95% CI, 4.0–6.1) with ARSI (HR, 1.02, 95% CI, 0.82–1.25, p = 0.55). At final OS analysis, median OS was 15.8 months (95% CI, 14.6–17.0) and 14.6 months (95% CI, 12.6–17.3), respectively (HR, 0.94, 95% CI, 0.77–1.14, p = 0.26). Patients receiving Pembrolizumab + Olaparib had higher ORR compared to men treated with ARSI (16.8% versus 5.9%). In addition, grade ⩾3 treatment-related AEs occurred in 34.6% and 9.0% of patients, respectively. 91
In the phase I/II MEDI4736 study (NCT02484404), 17 patients with mCRPC previously treated with Abiraterone or Enzalutamide were given Olaparib and Durvalumab. The primary endpoints were ORR and recommended dose for phase II (RP2D). The median rPFS was 16.1, with a 12-month rPFS of 51.5%. Patients showed AEs ⩾G3 including anemia (24%), lymphopenia (12%), infection (12%), and nausea (12%). 92 To date, the phase I/II QUEST trial (NCT03431350), testing the triple combination of Niraparib + Abiraterone with the anti-PD-1 agent Cetrelimab, is still ongoing.
Altogether, these clinical trials did not confirm the strong biological rationale suggesting a potential synergism between ICIs and PARPi in CRPC, although a modest activity of the combination was observed in HR deficient-PCa.
Crosstalk between AR and tumor immune-microenvironment in PCa
There is considerable crosstalk between AR and PCa TME resulting in immune suppression and tumor growth and spread. Preclinical evidence describes a usually ‘cold’ TME in PCa as it is characterized by a reduced T-cell infiltration, even though a subgroup of more immunogenic subsets of PCa has been observed. The latter are characterized by a higher PD-L1 expression and CD8+ T cell infiltration, with a better prognosis due to a lower risk of local and distant relapse. 93
AR signaling activation impacts on the elements of the innate immune response. The production of pro-inflammatory cytokines, such as IFN-γ, is reduced, impairing T cell activation. 94 In addition, AR promotes macrophage polarization toward the M2 subtype and inhibits the Toll-like receptor 4 pathway. A high infiltration of M2 macrophages has been associated with a higher risk of progression from localized to metastatic PCa. 95 In PCa TME, MDSCs, recruited with T-regs, maintain immune evasion and contribute to ADT resistance by IL-23 production. AR signaling also has effects on non-immune elements within the TME. It is involved in angiogenesis through the interaction with endothelial cells and expression of pro-angiogenic factors, such as vascular cell adhesion protein, NF-κB, vascular endothelial growth factor-α, TNF-α. 93
Androgens are described as suppressors of inflammation and immune function; after all, recently, the inhibition of AR signaling with ADT has been shown to restore the ability of CD8+ T cells to secrete pro-inflammatory cytokines and increase tumor responsiveness to immunotherapy. 94
The PCa triad among AR, DDR, and TME: implications for current practice and future perspectives
The AR remains a key target in PCa therapy and the continued development of new small molecules to target the AR signaling may improve future approaches and lend themselves to rational combination therapies, including with DDR-targeting agents or ICIs. These multiple interconnected pathways lead to potential combining treatments which can help overcome ARSI resistance and restores its sensitivity in PCa (Figure 5). Over the years, various studies have explored the critical roles of the AR, HRR machinery, and PARP in the pathogenesis of PCa, essentially revealing four mechanisms of action underlying the biology behind the combination therapy between PARPi + ARSI: (1) ARSI elicits a phenotype resembling HRR deficiency, (2) suppression of AR is linked with upregulated PARP activity, (3) PARP enhances AR function, and (4) PARPi may attenuate resistance to ARSI, especially due to BRCA2/Rb1 co-deletion, a driver event of disease progression. 96
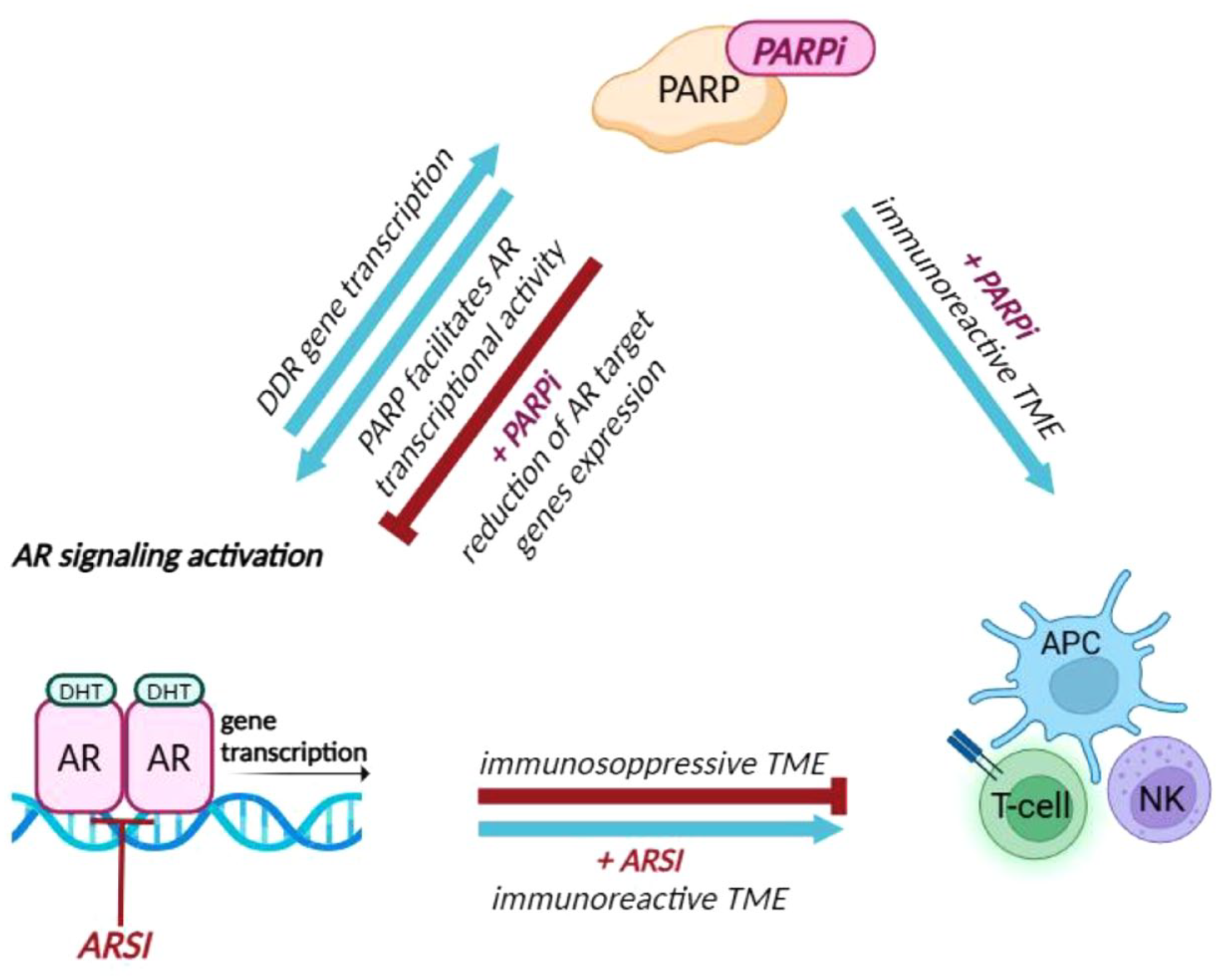
The ‘Perfect Triad’ among AR, DDR, and TME. AR regulates upstream transcription of several genes involved in the DDR pathway defining an ‘AR-associated DNA repair gene’ signature. In turn, DNA damage induces the activation of AR signaling. PARP-1, enzyme of the DDR complex, supports PCa growth and progression through AR transcriptional regulatory functions. Indeed, PARPi significantly reduces the expression of AR target genes mainly involved in PCa progression. Furthermore, PARPi interacts with the TME converting an immunologically ‘cold’ TME into a more immune-reactive one. PARPi enhances the recruitment of APCs and NK cells and leads to the activation of CD4+ and CD8+ T cells, resulting in activation of the immune response. In contrast, activation of AR signaling alters the balance between pro-inflammatory and anti-inflammatory cytokines impairing T cell recruitment creating an immunosuppressive TME, thus the use of ARSI could improve PCa immunogenicity.
However, despite growing evidence of preclinical studies pointing to a potentially broader applicability of PARPi combinations and clinical studies showing the outcome advantage to PARPi + ARSI treatment for HRR-positive mCRPC, some concerns need to be better explored about this therapeutic option, in particular, the optimal timing, the onset of important toxicities, which were similar to what had been observed in the second-line mCRPC studies, and their real impact on the QoL. 97
In the last years, among the several mechanisms underpinning the resistance to combining therapy with PARPi + ARSI, the interplay between hypoxia and androgen has been investigated to control a metabolic switch conferring resistance to ARSI 98 and, in addition, tumor hypoxia has been also associated with resistance PARPi and ICIs. For this reason, the association of these drugs with emerging therapeutic strategies, such as inhibiting of hypoxia inducibile factor 1 subunit alpha (HIF-1α) signaling or hypoxia-activated prodrugs, may offer additional insights to exceed the hypoxia barrier. 99
In addition, the identification of new targets, such as B7-H3 and MDSCs, integrated with the manipulation of the DDR, for example via PARP or ATR inhibitors or STING agonists, may determine a larger number of CRPC patients who can benefit from ICIs in future trials.
Conclusion
In conclusion, in the last decade preclinical and clinical evidence suggest that PARPi treatment alone or in combination with ARSI and/or other novel agents may confer benefits in PCa patients irrespective of specific biomarkers. While the knowledge of biomarkers in treatment selection for advanced PCa is growing, further data are warranted to provide comprehensive elucidation for guiding therapeutic decisions. Particularly, future research will be aimed at solving some essential questions: identifying the patient profile that will benefit most from these treatments, recognizing the best time to treat, expanding the use of genomic tests and optimizing combination therapies.