
Abstract
Aided by developments in diagnostics and therapeutics, healthcare is increasingly moving toward precision medicine, in which treatment is customized to each individual. We discuss the relevance of precision medicine in prostate cancer, including gene targets, therapeutics and resistance mechanisms. We foresee precision medicine becoming an integral component of prostate cancer management to increase response to therapy and prolong survival.
Keywords
Precision medicine in prostate cancer
Precision medicine is an emerging field that uses genetic and environmental markers to determine the diagnosis of disease subcategories, the prognosis of patients, the choice of therapeutic, and accurate dosing. Such practices have become increasingly sophisticated, involving information obtained from genomics, metabolomics, and proteomics. Moreover, precision medicine methodologies are now available throughout the space of medical oncology: drug design, drug development, use of currently approved medications, and salvage therapy.
Developments in the medical treatment of prostate cancer have historically depended on a more sophisticated understanding of how androgens influence the disease. In the 1940s, acting on observations from surgical castration, 1 Huggins and Hodges discovered that restricting testicular androgen production by means of oral estrogen analogues led to slower progression in many patients who developed metastatic disease.2,3 In the 1960s, several groups discovered the androgen receptor (AR),4–6 leading to the development of the first antiandrogens. 7 Agents used to counteract adrenal hyperplasias, ketoconazole, and aminoglutethimide8,9 were later repurposed to counteract prostate cancer in 1976 and 1983; these agents work by blocking the production of adrenal androgens. In the early 1980s, Andrew Schally developed the first luteinizing hormone releasing hormone (LHRH) agonists that were found to block testicular androgen production. 10 However, such therapies are only temporarily effective in patients with disease outside of the prostate gland, and the majority of patients develop castration-resistant prostate cancer (CRPC). Effective CRPC treatments have only been discovered in the past 13 years, when docetaxel chemotherapy was found to prolong overall survival.11,12 Nevertheless, treatment decisions were not typically based on the recognition of interindividual differences or a diversity of therapeutic options.
While a priori assessment of disease markers remains rare, recent advances in prostate cancer therapy have involved markers of prostate tumorigenesis, progression, and acquired therapeutic resistance (Table 1). Several of these markers are not involved in androgen pathways, reflecting a more nuanced understanding of the molecular drivers of prostate cancer. Such recognition of interindividual differences and a better understanding of the disease has led to several effective therapeutics that can be utilized in various disease subgroups harboring different genetic and mutational backgrounds. Prognostic tests have also recently emerged that can determine the likelihood of disease recurrence, metastases, and prostate cancer specific mortality (PCSM). The purpose of the present review is to discuss the recent progress of personalized approaches to the treatment of prostate cancer.
Genetic abnormalities in prostate cancer and potential therapies.
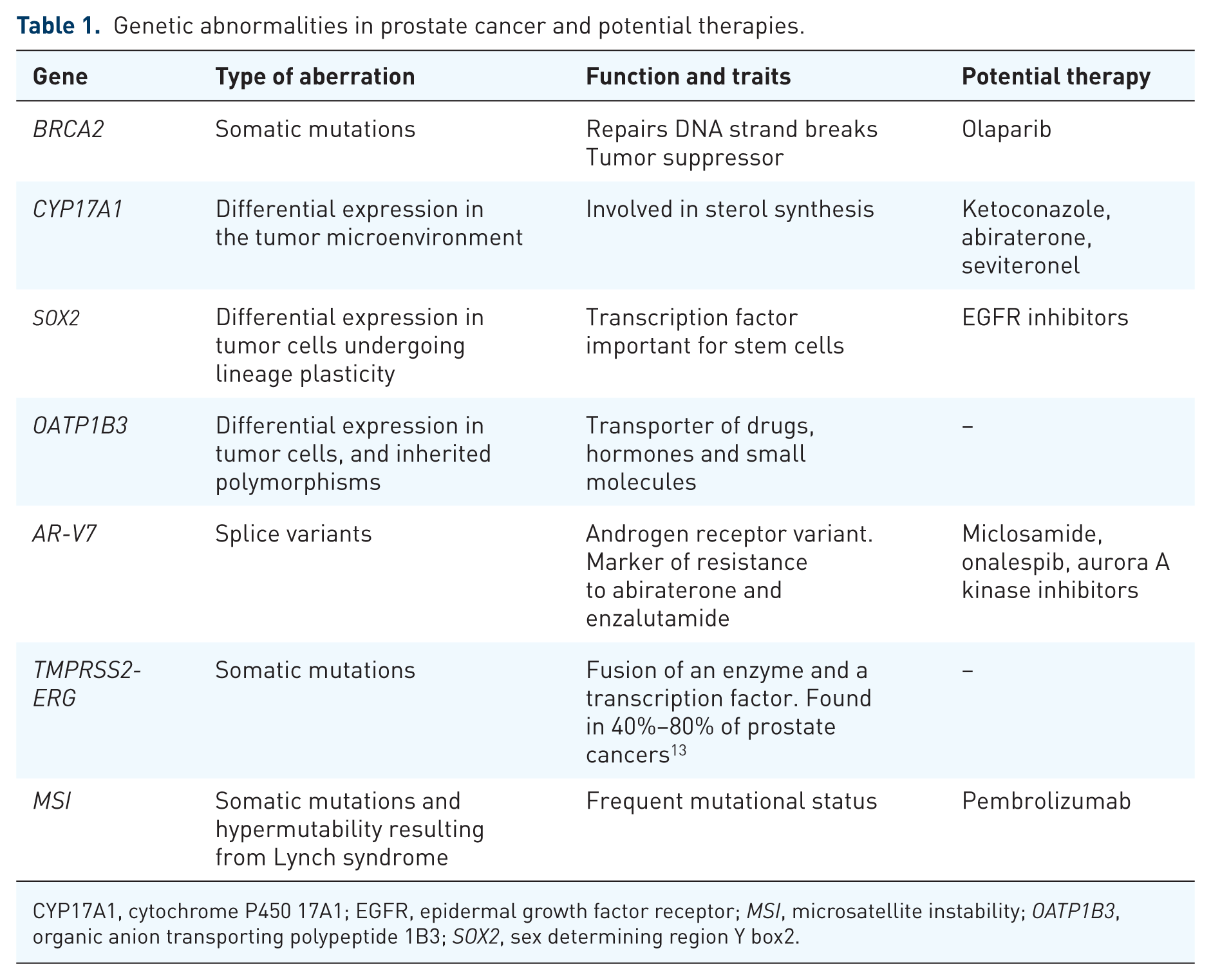
CYP17A1, cytochrome P450 17A1; EGFR, epidermal growth factor receptor; MSI, microsatellite instability; OATP1B3, organic anion transporting polypeptide 1B3; SOX2, sex determining region Y box2.
Selective therapeutics
Antiandrogens
There are currently several classes of antiandrogens. 14 Previously, antiandrogen therapy consisted of AR ligands that prevent AR transcription by blocking androgen binding (i.e. flutamide, nilutamide, and bicalutamide). In 603 treatment-naïve patients with disseminated prostate cancer, flutamide and leuprolide compared with placebo and leuprolide significantly prolonged duration of progression-free (16.5 versus 13.9 months) and overall (35.6 versus 28.3 months) survival. 15 In 457 patients initially treated with orchiectomy, nilutamide compared with placebo had a significantly higher proportion of patients with normal prostate-specific antigen (PSA) at 3 months (59% versus 28%) and longer progression-free (21.2 versus 14.7 months) and cancer-specific survival (37.0 versus 29.8 months). 16 In 205 patients with stage III or IV prostate cancer, bicalutamide with an LHRH agonist versus an LHRH agonist alone demonstrated a higher proportion of patients with normal PSA at 3 months (79.4% versus 38.6%) and a greater estimated 5-year overall survival rate (75.3% versus 63.4%).17,18
Newer antiandrogens typically also block AR transcription via multiple mechanisms. For instance, enzalutamide, apalutamide, and darolutamide are AR ligands which inhibit androgen binding, AR nuclear translocation, and the DNA-binding capacity of the AR. Niclosamide prevents AR binding to promoter sites on DNA and promotes AR proteolysis. EPI-001 and niphatenones both prevent AR-DNA binding. Because they target multiple elements of the androgen biosynthesis and gene expression pathways at once, newer antiandrogens inhibit intratumoral AR transcription more strongly than older antiandrogens and are typically more effective. For instance, 396 men with CRPC were treated with enzalutamide or bicalutamide with androgen deprivation therapy (ADT) in the double-blind, phase II STRIVE trial. 19 Patients treated with enzalutamide showed significantly better results than those with bicalutamide, including a proportion of patients with at least a 50% PSA decline (81% versus 31%), at least a 90% PSA decline (65% versus 9%), and progression-free survival (19.4 versus 5.7 months).
Androgen synthesis inhibitors
In order for the AR to adopt its active conformation, bind DNA, and subsequently activate effector genes, it must first bind either testosterone or its more preferred ligand, dihydrotestosterone (DHT). To prevent transcription of AR-downstream genes, which lead to cellular growth, various therapeutics target the androgen synthesis pathway in an effort to deplete the cells of potential AR ligands. The cytochrome P450 17A1 (CYP17A1) enzyme converts pregnenolone to 17α-hydroxypregnenolone by its hydroxylase activity and 17α-hydroxypregnenolone to dehydroepiandrosterone by its lyase activity; 17α-hydroxypregnenolone and dehydroepiandrosterone are important precursors for testosterone and DHT.
Therapeutics, such as abiraterone and ketoconazole, inhibit both reactions catalyzed by the CYP17A1 enzyme, while seviteronel selectively inhibits its lyase activity. Once testosterone is synthesized, the 5α-reductase enzyme converts it into DHT, which has a stronger affinity for the AR. Thus, androgen synthesis inhibitors have a place in prostate cancer treatment, and abiraterone is a standard of care for patients with metastatic, CRPC regardless of previous treatment with docetaxel.20,21 In 1195 patients with metastatic, CRPC previously treated with docetaxel, patients treated with abiraterone experienced a significantly longer overall survival than those treated with placebo (15.8 versus 11.2 months). In 1088 patients with metastatic, CRPC who had not received docetaxel, the abiraterone treatment group experienced a significantly longer radiographic progression-free survival than the placebo group (not reached versus 8.3 months) and a longer overall survival, even though not significant (35.3 versus 30.1 months). Abiraterone was also tested in 1199 patients with metastatic, castration-sensitive disease, and it showed significantly better results compared with placebo in radiographic progression-free (33.0 versus 14.8 months) and overall survival (not reached versus 34.7 months). 22 Unfortunately, while androgen synthesis inhibitors effectively stem tumor growth for a brief period of time, resistance to these therapies eventually develops, and prostate cancer can progress without androgen signaling.
Cytotoxic chemotherapy
Cytotoxic chemotherapy, such as docetaxel and cabazitaxel, remains a mainstay of prostate cancer treatment based on several studies. The TAX327 trial showed that in 1006 patients with metastatic, castration-resistant disease treated with mitoxantrone, docetaxel weekly or docetaxel every 3 weeks, overall survival favored the last group (16.5, 17.4 versus 18.9 months), so docetaxel every 3 weeks has become the standard. 11 Indication for docetaxel was expanded to include metastatic, castration-sensitive disease based on two trials, STAMPEDE and CHAARTED.23,24 In the STAMPEDE trial with 2962 patients, the group treated with docetaxel and ADT with or without radiotherapy exhibited a significantly longer overall survival compared with the group treated with ADT with or without radiotherapy (81 versus 71 months). This finding was duplicated in the CHAARTED trial with 790 patients, in which patients treated with docetaxel and ADT showed a significantly longer overall survival compared with those treated with ADT (57.6 versus 44 months). For patients with metastatic, castration-resistant disease already treated with docetaxel, cabazitaxel was shown to be efficacious. 25 In 755 patients treated with either cabazitaxel or mitoxantrone, the former group showed a significantly longer overall survival (15.1 versus 12.7 months). Thus, docetaxel and cabazitaxel are standards of care in metastatic prostate cancer.
Previous dogma stated that the taxanes acted by inducing G2/M cell cycle arrest. 26 Yet, several studies have now demonstrated that docetaxel and cabazitaxel also disrupt microtubules that are required for AR nuclear translocation, and that docetaxel is a substrate for dysregulated transporters that are expressed de novo in prostate cancer. 14 Thus, even the application of docetaxel and cabazitaxel is warranted in some genetic contexts and not others.
Targeting AR cofactors
In addition to blocking androgen biosynthesis and AR gene signaling, drugs have recently been developed to target transcriptional coactivators and epigenetic regulators of AR. One such example is the bromodomain and extraterminal (BET) inhibitor, JQ1. JQ1 works by inhibiting the two bromodomains in BRD4 that are essential for its binding to the AR. Because BRD4 is the factor that helps the AR recruit RNA polymerase II to gene promoters, inhibition of BRD4 with JQ1 results in a loss of AR-driven gene transcription. In a VCaP xenograft model, enzalutamide demonstrated a prometastatic effect that was not seen with JQ1 treatment. JQ1 also inhibited tumor growth and AR target gene expression more than enzalutamide. However, JQ1 only altered gene expression in AR-positive cell lines, indicating that JQ1 treatment would be the most effective earlier in prostate cancer before patients’ tumors progressed to castration resistance. 27 While this treatment has yet to be tested in a clinical setting, these in vitro and in vivo results heavily suggest that AR cofactors and transcriptional regulators pose attractive targets for therapeutic development.
Therapeutic options for gene dysregulation in prostate cancer
ADT resistance
AR splice variants
While numerous AR splice variants have been identified, AR-V1 to AR-V7 are spliced in such a way that truncates the AR’s ligand binding domain and confers the potential for constitutive activation of the AR. However, AR-V2, AR-V3, and AR-V4 are not observed in clinical prostate cancer.28,29 AR-V1 is expressed more frequently in CRPC, but it lacks several amino acids that decrease its ability to localize to the nucleus, and its expression is not correlated with treatment outcome in patients.28,29 The AR-V7 splice variant excludes the ligand binding domain, making it the most relevant AR splice variant in prostate cancer. This deletion results in a constitutively active AR that can translocate to the nucleus and signal gene expression even in the absence of testosterone or DHT.14,28,29 The upregulation of the AR-V7 splice variant likely occurs due to the increased expression of various splicing factors, such as splicing factor proline and glutamine-rich (SFPQ), that is observed in prostate cancer.30–32 However, as of yet there are no clinically approved treatments targeting these factors.
Patients with AR-V7 experienced lower response rates and survival after treatment with enzalutamide and abiraterone compared with those without the variant.33,34 Patients with AR-V7 had better response to and survival with taxanes than with abiraterone and enzalutamide. These phenomena are attributable to an absence of the ligand-binding domain. Such differences in response and survival by treatment type did not occur for patients lacking AR-V7. Therefore, AR-V7 may be a useful biomarker for prostate cancer treatment selection.
Candidate agents, which may target AR-V7, include niclosamide, onalespib, and aurora A kinase inhibitors. Niclosamide is an antihelminthic drug, which reduces AR-V7 protein levels and inhibits growth of AR-V7-expressing cancer cells in vitro and in vivo. 35 It also shows synergistic effects with enzalutamide or abiraterone in vitro and in vivo.35,36 Onalespib, a heat shock protein inhibitor, reduces the generation of AR-V7 by introducing alternative splicing events. 37 Aurora A kinase inhibition reduces AR-V7 mRNA and protein levels. 38 In summary, specific agents may soon become available for patients for whom abiraterone and enzalutamide are ineffective.
ADT evasion via CYP17A1 androgen synthesis
In normal prostate cells, AR-mediated gene signaling is activated by gonadally derived testosterone, which is subsequently converted into the more potent androgen, DHT. However, in prostate cancer, tumors utilize the CYP17A1 enzyme to convert androgen precursors from the adrenal glands, such as dehydroepiandrostenedione (DHEA), into testosterone, thereby evading the low androgen environment caused by chemical or surgical castration. The CYP17A1 enzyme specifically catalyzes the synthesis of androgen and glucocorticoid precursors (17α-hydroxypregnenolone and 17α-hydroxyprogesterone) through hydroxylase activity and weak androgens (DHEA and androstenedione) via lyase activity. 39 Once these precursors are converted to testosterone, the steroid-5α-reductase isoenzyme-2 (SRD5A2) reduces testosterone to DHT, which has an even higher affinity for the AR than testosterone and, subsequently, plays a larger role in upregulating androgen-dependent gene transcription. 40 While SRD5A2 is the dominant isozyme in normal prostate tissue, prostate tumors upregulate the SRD5A1 enzyme.
As a result of its role in developing resistance to ADT, CYP17A1 is an established target for prostate cancer therapy. Ketoconazole, an antifungal, weakly and nonspecifically inhibits CYP17A1 and has been noted to be active in prostate cancer at high doses. 9 A newer agent, abiraterone, more selectively inhibits CYP17A1 resulting in decreases in circulating androgens (DHEA, testosterone, and dihydrotestosterone). Thus, abiraterone inhibits AR signaling, prolongs survival, and is a standard therapy for metastatic, castration-resistant cancer.41,42 Since abiraterone inhibits CYP17A1 hydroxylase sixfold more selectively than CYP17A1 lyase, it causes glucocorticoid deficiency and must be administered with prednisone. Another CYP17A1 inhibitor, seviteronel, more selectively inhibits the lyase function of CYP17A1 and thereby reduces androgen biosynthesis, AR signaling, and tumor growth without causing glucocorticoid deficiency. Therefore, should seviteronel demonstrate similar or superior efficacy to abiraterone, it may also become a standard option.43,44
OATP1B3
The organic anion transporting polypeptide (OATP) family of transporters is implicated in prostate tumorigenesis, with relevant potential therapy to be developed. As indicated by their name, the OATP family of transporters carries drugs, hormones including testosterone, and small molecules, and thus is essential for a functional liver, which processes and detoxifies numerous substances. 45 OATP family expression has also been noted in other organs, such as breast, lung, and prostate, and specifically OATP1B3, a member of this family, has been implicated in prostate tumorigenesis. 46 Testosterone uptake differed among the cells exhibiting different polymorphisms, and the patients who harbored the haplotypes with reduced testosterone uptake exhibited longer progression-free and overall survival than those who did not. Furthermore, OATP1B3 mRNA and protein expression directly correlated with Gleason scores, implicating it as a biomarker of aggressive disease. 47 Therefore, OATP1B3 inhibition leading to testosterone reduction may have a role in prostate cancer therapy.
It was also recently determined that in addition to testosterone, OATP1B3 has the ability to transport docetaxel and abiraterone into the cell.48,49 The knowledge that OATP1B3 is upregulated in resistance can allow physicians to make more informed decisions regarding the sequence of taxane and androgen biosynthesis targeting drugs.
Therapeutic options for somatic mutations in prostate cancer
AR mutations
As tumors grow and progress, they accumulate somatic mutations that benefit the tumor. Because the AR plays a large role in the development of prostate cancer, mutations that allow the AR to bind alternative ligands, such as estrogen and progesterone, and continue to signal gene expression in the absence of androgens are selected for. Among the more than 70 different missense mutations identified in prostate cancer, H874Y, F876L, T877A, and W741L/C are particularly interesting because they are linked to drug resistance and disease progression.14,50,51
Each of these mutations changes the conformation of the AR by replacing amino acids in the ligand binding pocket, so that it can bind ligands other than testosterone and DHT. The H874Y and T877A somatic mutations similarly affect the AR and create more space in the ligand binding pocket, allowing the AR to bind estradiol, progestins, and cyproterone acetate, among other ligands.14,52,53
The F876L and W741L/C mutations relate more directly to drug resistance. The F876L mutation alters the ligand binding pocket so that enzalutamide binds the AR and promotes its activation rather than inhibition. 54 Similarly, the W741L/C mutation promotes bicalutamide binding in a way which moves the AR into its active conformation.55,56 Such mutations create obstacles for designing effective treatment of CRPC.
TMPRSS/ERG
An important discovery was noted in 2005, in which TMPRSS2-ETS gene fusions were noted in 23 of 29 prostate cancer tissues, identifying it as an important somatic mutation. 57 This discovery was notable because such gene translocations and consequent fusions had been well described for other malignancies, which include BCR-ABL in chronic myeloid leukemia, MYC-IGH in Burkitt’s lymphoma, and EWS-FLI1 in Ewing’s sarcoma but not for prostate cancer.58–60 As a result of the gene fusions, TMPRSS2 may mediate the overexpression of ETS family members, ERG or ETV1. This is significant because ERG is a frequently expressed oncogene in prostate cancer, and TMPRSS2-ERG fusions mediate invasion in both in vitro and in vivo models of prostate cancer.61,62 When coupled to serum PSA, urine detection of TMPRSS2-ERG has been shown to enhance the ability to predict prostate cancer in tissue biopsy. 63 However, TMPRSS2-ERG has been an elusive therapeutic target, with mixed results from clinical trials of agents including inhibitors of poly ADP ribose polymerase 1 (PARP1), DNA protein kinase, and histone deacetylase 1. 64 Nevertheless, the potential of a successful, targeted therapy for a majority of prostate cancers has sustained research efforts into TMPRSS2-ERG.
BRCA1/2
BRCA1 and BRCA2 mutations have emerged as potential therapeutic targets in certain patients with prostate cancer because cells expressing such mutations have a significantly diminished capacity to repair double-stranded DNA (dsDNA) breaks. Studies have reported on the frequencies and relative importance of BRCA1/2 mutations. In 692 men with metastatic disease, 0.9% had germline mutations in BRCA1 and 5.3% in BRCA2. 65 In 251 Ashkenazis with prostate cancer, 2% had BRCA1 mutations compared with 3.2% with BRCA2. 66 BRCA1 mutation carriers had no increased risk of prostate cancer, in contrast to a 4.8-fold increased risk among BRCA2 mutation carriers.
Since normal (BRCA1/2-positive) cells efficiently repair dsDNA breaks, tumor cells that have acquired BRCA1/2 mutations can be specifically targeted.67,68 Olaparib, a PARP inhibitor, prevents repair of single-stranded DNA (ssDNA) breaks, leading to rapid accumulation of dsDNA breaks in BRCA1/2 mutated cells. A phase I study showed that not only did olaparib have a favorable side-effect profile, it also showed antitumor activity only in those with BRCA1 or BRCA2 mutation. 69 The TOPARP-A trial assessed olaparib as a therapy among 50 patients with metastatic, CRPC, most of whom were pretreated with abiraterone (96%), androgen deprivation, and docetaxel (100% each). 70 In TOPARP-A, the 16 responders included all seven patients with a BRCA2 mutation. Thus, olaparib may become part of the prostate cancer therapeutic regimen for a subset of patients with DNA repair defects. Of interest, circulating cell-free DNA (cfDNA) samples collected during the TOPARP-A trial showed great promise for precision medicine as a prognostic, predictive, and resistance biomarker. 71 Reduction in their levels correlated with overall survival. Not only were DNA repair mutations detectable in cfDNA, their allelic frequencies decreased in responders, and cfDNA could be utilized to detect emergence of resistance to therapy.
Microsatellite instability
The US Food and Drug Administration approved the use of pembrolizumab in patients harboring tumors with microsatellite instability (MSI) regardless of the tumor site. 72 A minority of prostate tumors (~4–25%) demonstrate MSI, 73 which is defined by hypermutability of repeated sequences of DNA resulting from mismatch repair deficiencies and leads to heritable risk of prostate cancer.74,75 Such mutational burden results in sensitivity to blockade of the programmed cell death protein 1 (PD-1), a negative feedback pathway that represses T helper 1 (Th1) cytotoxic immune response. 76
Therapeutic resistance mechanisms
ARv7
Since Antonarakis and colleagues linked the presence of AR-V7 mRNA in circulating tumor cells with resistance to both enzalutamide and abiraterone, AR-V7 has been at the forefront of prostate cancer resistance research. 77 As mentioned previously, the AR-V7 splice variant excludes the AR’s ligand binding domain, which is the target of both enzalutamide and (indirectly) abiraterone. Without the ligand binding domain, there is no place for enzalutamide to bind and, as a result, AR-V7 continues to translocate to the nucleus and activate AR-dependent gene transcription. Abiraterone works by inhibiting the synthesis of both testosterone and DHT, however the AR-V7 isoform no longer depends on ligand binding for activation, and therefore the therapeutic activity of abiraterone no longer has an effect.
Additionally, a growing body of evidence suggests that AR-V7 may play a role in an overlapping resistance mechanism to both abiraterone and docetaxel. In a clinical study, patients who received docetaxel treatment following abiraterone did not respond as well to docetaxel as those who had not received prior abiraterone treatment. 78 Furthermore, patients who were abiraterone resistant also demonstrated resistance to subsequent docetaxel treatment.
Sex determining region Y box2
Sex determining region Y box2 (SOX2) is an essential stem cell transcription factor that promotes enzalutimide resistance by promoting lineage plasticity, a process by which prostate tumor cells undergo dedifferentiation to neuroendocrine cells that simultaneously display luminal, basal, and epithelial characteristics. 79 SOX2 expression also promotes growth and inhibits apoptosis in untreated CRPC cells,80,81 and its expression is clinically associated with biochemical recurrence, lymph node metastasis, migration, and invasion of prostate cancer cells. 82 Although no SOX2 inhibitor for prostate cancer therapy has been developed, an epidermal growth factor receptor (EGFR) inhibitor reduces SOX2 expression in prostate cancer cells, implicating a possible therapeutic option. 83
Prognostic genetic testing
Integration of genomics and pharmacogenomics into patient care is closely connected with improvement of diagnostics, and RNA profiling tests, such as Decipher(GenomeDx, San Diego, CA), Oncotype DX (Genomic Health, Inc., Redwood City, CA), and Prolaris (Myriad Genetic Laboratories, Inc., Salt Lake City, UT), can offer additional information for providers and patients with prostate cancer (Table 2). Decipher predicts metastasis and prostate-cancer-specific mortality after radical prostatectomy.84,85 Oncotype DX predicts biochemical recurrence after radical prostatectomy and prostate-cancer-specific mortality in patients under active surveillance.86,87 Prolaris predicts biochemical recurrence, metastasis, and prostate-cancer-specific mortality after radical prostatectomy, as well as mortality in those under active surveillance.88–90 How these tests will fare compared with one another remains to be determined. Although they are currently not part of treatment planning for prostate cancer, they may become so in the future. Unfortunately, Decipher, Oncotype DX, and Prolaris provide prognostic, but not predictive information, and they are costly (greater than $3000 per test). 91 The lack of predictive information deters widespread adoption, and a 24-gene signature, reported to predict which patients with prostate cancer would benefit the most from postoperative radiation, may address the deficiency. 92 A retrospective study noted that patients with high Post-Operative Radiation Therapy Outcomes Score (PORTOS (Genome Dx, San Diego, CA)) who had postoperative radiotherapy were less likely to have metastasis at 10 years compared with those with high PORTOS who did not. After PORTOS is validated in additional trials and patients, and becomes commercially available, it may become a useful tool in prostate cancer management.
Genomic diagnostic tests for prostate cancer.
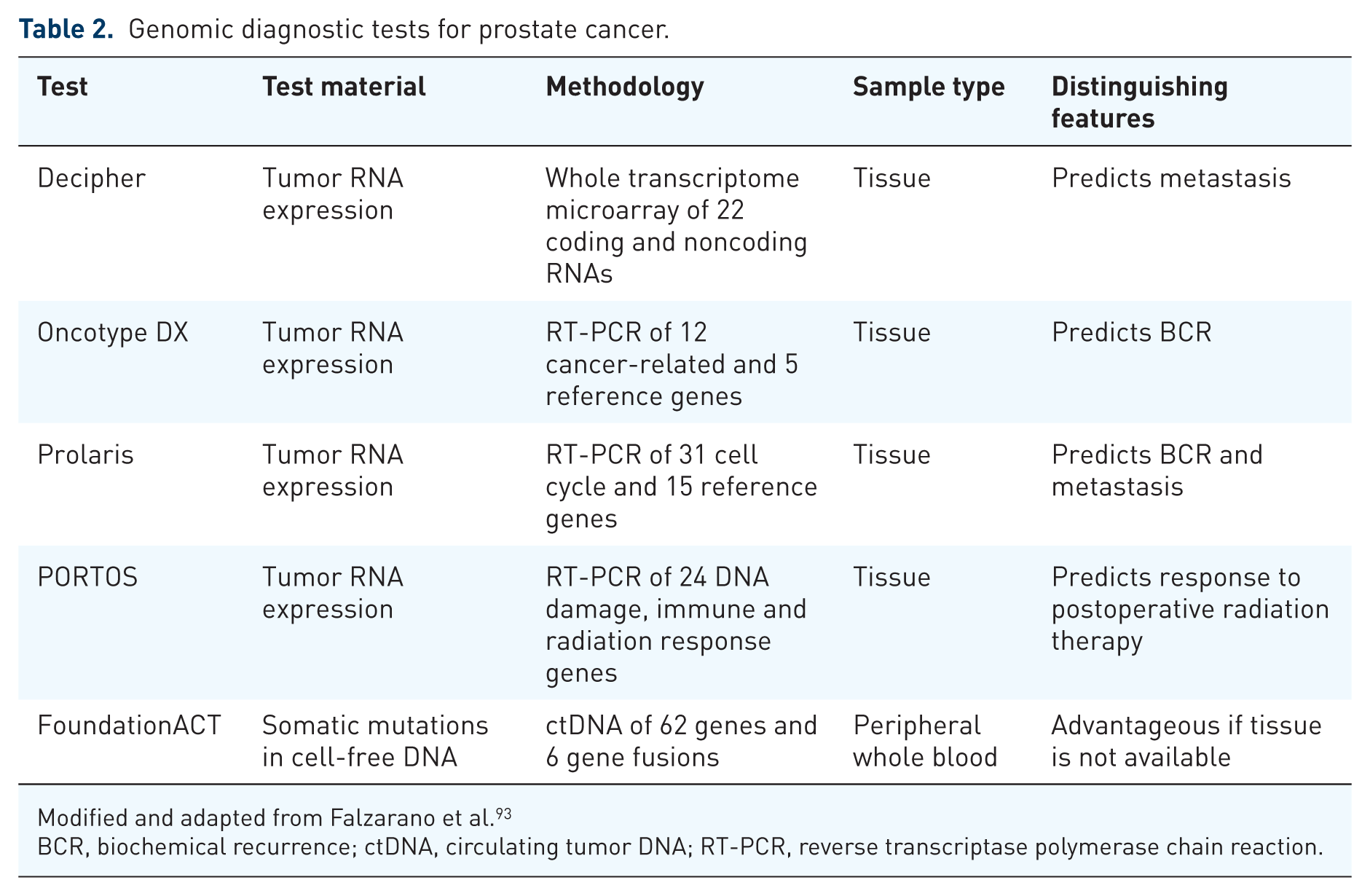
Modified and adapted from Falzarano et al. 93
BCR, biochemical recurrence; ctDNA, circulating tumor DNA; RT-PCR, reverse transcriptase polymerase chain reaction.
Another issue facing pharmacogenomics is utilization of information from diagnostic tests. Several companies, including Foundation Medicine (Cambridge, MA), employ high-throughput sequencing to detect germline or somatically acquired genomic perturbations in patients with solid and hematological malignancies.94–96 Personalized, targeted treatments based on the genomic information thus acquired have led to mixed results, ranging from no difference in outcome to improved response rates in metastatic breast cancer. This spectrum of responses affirms that genomic aberrations and a multitude of other factors influence treatment response, and furthermore, survival. In addition, understanding of disease mechanism and treatment resistance needs to improve to take advantage of genomic diagnostic information. Foundation Medicine offers several tests, including FoundationACT, which is designed to detect mutations in cancers of breast, colon, lung, and prostate. Hopefully, FoundationACT and other genomic diagnostic tests will yield discoveries, which will ultimately lead to improved responses in prostate cancer.
Future of precision medicine in prostate cancer
As outlined so far, successful establishment of precision medicine in prostate cancer hinges on numerous factors, including accurate diagnostic tests, specific gene targets, and effective treatments. In order to put precision medicine into practice, clinicians and scientists need to be able to readily access clinical and genomic data of patients, and some search tools include Oncology Data Retrieval Systems (OncDRS (Dana Farber Cancer Institute, Cambridge, MA)) and GeneMed (National Cancer Institute, Bethesda, MD).97,98 Physicians can utilize OncDRS to quickly search and obtain clinical and genomic data of patients with cancer, and such information can be used to tailor treatment and design clinical trials. Within a year of release, OncDRS aided data retrieval for more than 50 publications. GeneMed has facilitated a molecular profiling-based assignment of cancer therapy (MPACT) clinical trial [ClinicalTrials.gov identifier: NCT01827384] by operating as a hub to integrate information from biostatisticians, clinicians, and sequencing laboratory. This trial uses next-generation sequencing to identify actionable mutations in patients with advanced solid tumors and randomize eligible patients to two arms, one with a targeted drug for mutation, and the other with a nontargeted drug. The list of targeted drugs includes inhibitors of MEK, mTOR, PARP, and Wee1. Upon disease progression, patients on the arm with the nontargeted drug can cross over to the arm with the targeted drug, and outcome measures include response rate and progression-free survival. At the time of writing the trial is ongoing and recruiting participants [ClinicalTrials.gov identifier: NCT01827384].
Overall, precision medicine is not yet standard practice in prostate cancer, so more work is needed. Unlike BRCA2 and CYP17A1, SOX2, OATP1B3, AR-V7, and TMPRSS2-ERG currently do not have approved targeted agents. It also appears that therapeutics such as pembrolizumab are effective in certain tumors with high microsatellite instability or mismatch repair deficiencies.99,100 Thus, detection of genomic abnormalities with appropriate treatments may help establish precision medicine in prostate cancer. In addition, results from genomic diagnostic tests, such as Decipher, Oncotype DX, Prolaris, PORTOS, and FoundationACT need to be interpreted with caution because no large randomized clinical trial has supported their routine use in patient care. Another reason to apply caution is that a considerable variation of gene expression levels exists among tissue cores from the same patients, so multiple biopsies from the same patients may need to be subjected to genomic diagnostic tests, and their results averaged before application. 101 Although FoundationACT reduces sampling bias by using peripheral blood, it, too, has not been validated in a large prospective study. Also, scores from Decipher, Oncotype DX, and Prolaris varied both within and among tissue cores from the same patients, which indicates a need for a uniform, standardized genomic diagnostic test in prostate cancer. Despite these concerns, the field is moving toward precision medicine aided by advances in diagnostics, therapeutics, understanding of disease mechanism, and integration of clinical and genomic data (Figure 1). Therefore, clinicians and scientists are well positioned to take advantage of the new resources to establish precision medicine in prostate cancer.

Components of precision medicine cooperating towards a better outcome (dx: diagnostic).
Footnotes
Acknowledgements
Edel M. McCrea and Daniel K. Lee are joint first authors.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.