
Abstract
In spite of decades of research, cancer survival has increased only modestly. This is because most research is based on models of primary tumors. Slow recognition has begun that disseminated, dormant cancer cells (micrometastatic cells) that are generally resistant to chemotherapy are the culprits in recurrence, and until these are targeted effectively we can expect only slow progress in increasing overall survival from cancer. This paper reviews efforts to understand the mechanisms by which cancer cells can become dormant, and thereby identify potential targets and drugs either on the market or in clinical trials that purport to prevent metastasis. This review targets the most recent literature because several excellent reviews have covered the literature from more than two years ago. The paper also describes recent work in the authors’ laboratories to develop a screening-based approach that does not require understanding of mechanisms of action or the molecular target. Success of this approach shows that targeting micrometastatic cells is definitely feasible.
Introduction
The War on Cancer was declared by President Richard M. Nixon in 1974, and in the ensuing 40 years, billions of dollars have been spent on it. Much has been learned about the mechanisms of cancer growth and progression; genes that are responsible for cancer have been identified and many of these proteins coded for by cancer genes have been targeted with specific agents. Yet, remarkably, survival figures have changed relatively little [Milojkovic and Apperley, 2009]. People are living longer with fewer side effects from surgery, radiation and chemotherapy, but in the end, with only a few exceptions, cancer remains an incurable disease unless it is eliminated by surgery. In large part, this occurs because so much of drug development is based on models that may not represent the natural history of human cancer [Leaf, 2004]. Most research uses models of primary tumors, and yet over 90% of people who die of cancer do not die from their primary tumor but rather, largely, from drug-resistant metastatic tumors [Talmadge and Fidler, 2010]. The successes that have been achieved are from use of drugs that happen to target metastatic tumors as well as the primary, but even with modern, targeted therapies, the main effect is often to delay progression or recurrence. For example, a recent study comparing the combination of trastuzumab and lapatinib when compared with lapatinib alone in patients with progression of metastatic breast cancer on prior trastuzumab-containing therapy, showed an improvement in median progression-free survival from 8.1 weeks to 12.0 weeks and by 32 weeks, the Kaplan–Meier curves had converged at under 20% survival [Swain et al. 2015]. A recent review of adjuvant and neoadjuvant therapy in muscle-invasive bladder cancer reported that only a few trials showed a significant effect, and most showed none [Balar and Milowsky, 2015]. The most notable success in preventing recurrence has come with hormonally sensitive cancers such as estrogen receptor-positive breast cancers for which continuing tamoxifen for five years reduces recurrence significantly. It was further found that 10 years of therapy with tamoxifen adds additional benefit, suggesting that nascent tumors can be maintained in a dormant state [Davies et al. 2013]. However, the effect was still relatively modest; the cumulative risk of recurrence during years 5–14 was 21.4% for women allocated to continue versus 25.1% for controls; breast cancer mortality during years 5–14 was 12.2% for women allocated to continue versus 15.0% for controls (absolute mortality reduction, 2.8%). The goal of this perspective-type review is to provide an overview of the most recent literature and describe a new screening-based approach to identify new compounds for targeting dormant cells that demonstrates targeting disseminated micrometastatic cells is feasible.
Targeting metastases versus disseminated micrometastatic cells
The lack of drugs to target metastases arises because of a lack of a mechanistic understanding of metastasis and a lack of models for screening for new drugs that target metastases [Weber, 2013]. Nor have mechanistic studies to date provided a clear strategy for how to target metastases, with the exception of a few cancer types. Weber outlined two strategies for developing drugs to treat metastatic disease [Weber, 2013]: the first is to develop drugs that prevent dissemination of cancer cells and would be administered immediately upon diagnosis, and the second is to develop drugs that target pre-existing metastatic tumors. Unfortunately, both of these strategies are likely to have only limited efficacy. First, as shown by several studies and the number of patients who are apparently ‘cured’ only to develop metastases months, years, or even decades later, in far too many cases, dissemination occurs early, even before initial diagnosis [Melchior et al. 1997; Gray, 2003]. Second, macroscopic metastatic tumors will contain millions of cells, and tumor heterogeneity that so far has foiled most efforts to cure cancer, and so will remain a factor. Finally, although metastases are formed from cells derived from the primary tumor, they likely originate from very rare cells. Talmadge and Fidler estimate that no more than 0.01–0.1% of cells in a primary tumor are capable of establishing metastases [Talmadge and Fidler, 2010]. Whether these are so called cancer stem cells is not known and currently is a topic of debate [Sun and Ma, 2015].
We suggest that the most vulnerable and the rate-limiting step in metastasis is not dissemination but rather the escape of micrometastatic cells, single cells and small clumps of cells, from the suppressive effects of multiple mechanisms that keep them dormant after seeding at this secondary site. At this stage, the potential metastases are single or small clusters of cells lacking a clear blood supply and only a limited number of them are disseminated through the patient’s body. Targeting such micrometastatic cells might avoid many of the problems of tumor heterogeneity, as micrometastases have only a small number of cells and as such would be less heterogeneous than would a macroscopic tumor. In order to target such micrometastatic cells, the mechanism for their dormancy and reactivation should be known and is a huge area of active research. The clinical potential for such therapy is high, with the initial target population being patients who are apparently cancer free following definitive surgical or radiological treatment, but who still have a significant probability of recurrence.
Several mechanisms have been identified by which disseminated cancer cells or small tumors can remain dormant [Almog, 2010; Osisami and Keller, 2013]. Almog distinguished three mechanisms: angiogenic insufficiency, immunosurveillance and exiting the cell cycle due to host-specific features at the metastatic site or selection by chemotherapy [Almog, 2010]. Osisami and colleagues provided a similar, but slightly different list: tumor microenvironment factors such as cytokine expression, immunosurveillance and angiogenic, metastasis suppressor-gene activity and cancer therapeutics that select for cells that have exited the cell cycle [Osisami and Keller, 2013]. Angiogenic insufficiency occurs when a cluster of cells is unable to recruit a blood supply to support inexorable growth. Cells may be proliferating rapidly, but the lack of nutrients leads to a balance between death and proliferation. Immunosurveillance can also prevent inexorable growth but not to the level to completely extinguish the microtumor. The immune system can also play a more direct role in maintaining dormancy. Antibodies directed against the immunoglobulin receptor of a B-cell lymphoma model induced dormancy over an extended time [Rabinovsky et al. 2007]. There are multiple rationales for cells to exit the cell cycle, including treatment with chemotherapy [Osisami and Keller, 2013].
One mechanism of dormancy that is oftentimes overlooked is the suppression of malignancy by the normal extracellular matrix (ECM). Iozzo demonstrated over 20 years ago that the stroma contains both agonistic and antagonistic signaling [Iozzo, 1995], and the antagonistic elements could keep micrometastatic cells in a nondividing state. If these micrometastatic cells are not dividing actively, they will generally be resistant to chemotherapy because most chemotherapeutic agents target dividing cells. The function of the stroma is to provide signals to maintain the overlying epithelium in a differentiated state and to carefully regulate replication appropriate to the particular tissue. These signals are inherently unfriendly to cancer, and it is not unreasonable to expect that single disseminated tumor cells would be sensitive to the suppressive effect of the normal ECM, given that malignant growth is accompanied by extensive remodeling of the local stromal environment to be more ‘cancer friendly.’
Models of dormancy
Numerous in vitro and in vivo models have been developed to recapitulate aspects of dormancy and the influence of stromal microenvironment. More recent models usually not only include the metastatic cancer cells, but also contain the ECM, stromal cells, and ideally, even immune cells, to most accurately mimic the complex interactions between cancer cells and the metastatic microenvironment. Among the ECM scaffolds or extracts used are poly (ε-caprolactone), fibronectin, collagen, and basement membrane extract (Table 1). Work has begun to unravel the complex agonistic and antagonistic elements in stromal signaling. Barkan and colleagues [Barkan et al. 2008] engineered breast cancer cell lines to express a dormant phenotype, but this only emerged when the cells were grown in three dimensions. The transition from quiescence to proliferation of one cell line was dependent on fibronectin production and signaling through integrin β1, that led to reorganization of the cytoskeleton and formation of F-actin stress fibers [Barkan et al. 2008]. It has also been described that the ECM alone can induce a permanent or temporary state of dormancy in cancer cells [Barkan et al. 2010]. Heparanase appears to be involved in remodeling the local ECM and could represent a mechanism by which micrometastatic cells eventually escape the suppressive effects of the normal ECM [Cohen et al. 1994; Gotte and Yip, 2006; Caruana et al. 2015]. Novel 3D organotypic culture systems have been emerging to address the complexity of the microenvironment (Table 1). A combination of cancer cell lines and primary human cells are cocultured to establish the 3D culture system. The noncancerous cells consist of a variety of cell types based on the microenvironment that is being mimicked (e.g. mesenchymal cells, fibroblasts, bone marrow cells, osteoblasts). The involvement of microvasculature or fibrous stroma in inducing and maintaining dormancy can be studied in these coculture models with endothelial cells or fibroblasts, respectively. An ex vivo model has been established to assess the metastatic progression of single cells [Mendoza et al. 2010]. In this model, tumor cells are injected into a mouse and the lungs removed and sliced for culturing and monitoring.
In vitro models for studying dormancy.

ECM, extracellular matrix; 2D, 2 dimensional; 3D, 3 dimensional; ER, estrogen receptor.
In vitro models are necessary for initial screening assays, but follow-up studies necessitate the use of animal models to validate the findings. Ideally, immunocompetent mice should be used since the importance of the immune system in the microenvironment and dormancy has been widely recognized [Manjili, 2014]. Nonetheless, human cancer-cell implantation into immune-deficient animals can also yield valuable information about metastasis and dormancy. Several models involve the generation of metastatic cancer sublines from cell lines or primary-tumor tissue that when implanted subcutaneously or orthotopically into animals form organ-specific metastases [Izraely et al. 2011; Sakamoto et al. 2015] (Table 2). Genetically engineered mouse models (GEMM) of oncogene (e.g. Kras, c-myc) ablation or transgenic mice (e.g. MMTV-PyMT) that spontaneously develop tumors and metastases, can yield a longer window of dormancy suitable for studying the process of reactivation or efficacy of metastasis-prevention agents, as tumor-implantation models are often fast progressing. Orthotopic implantation of cancer cells with subsequent resection of the primary tumor has also been shown to yield a period of dormancy followed by reactivation [Marshall et al. 2012]. These resection models not only may help in the understanding of signaling pathways of dormancy, but also offer a platform to test potential agents targeting fully formed metastases. In general, all of these models only reflect components of the complex process of dormancy and possibly metastatic progression, but can yield clinically relevant data if used with an understanding of their positive attributes and limitations.
In vivo dormancy/metastasis mouse models.

New drugs used for tumor recurrence
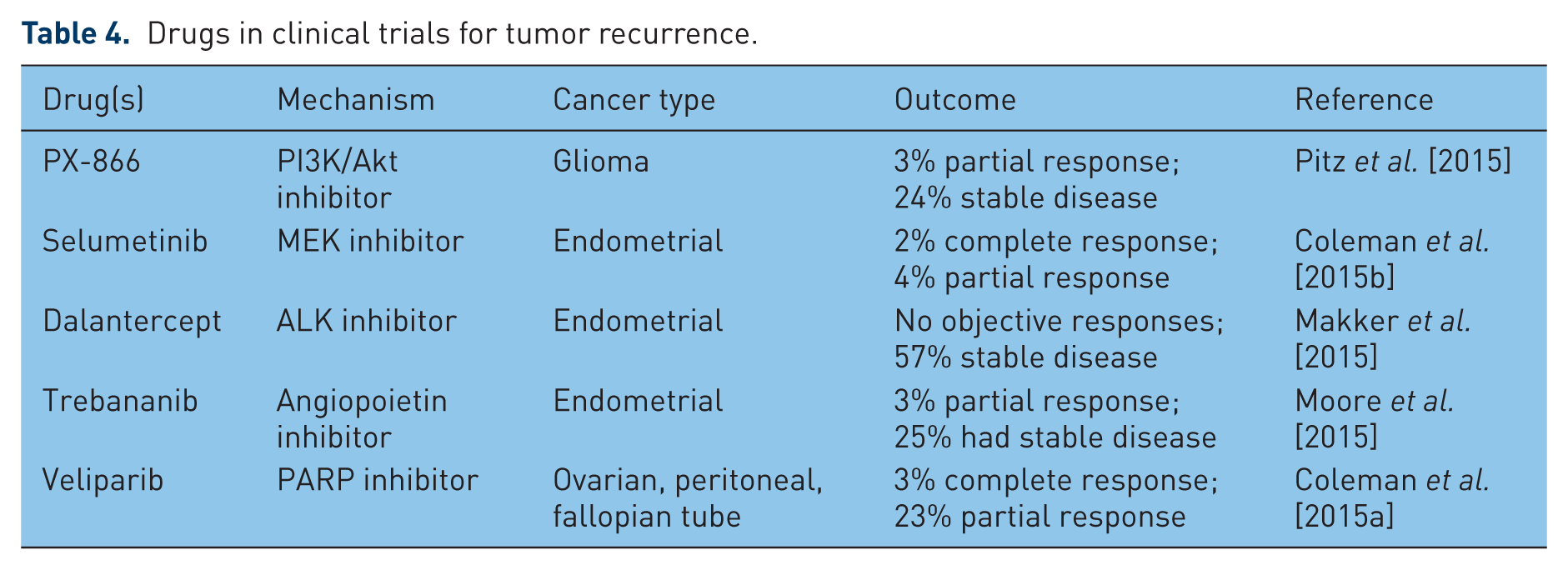
Most of the drugs approved for treating recurrent cancer are the same drugs used to treat primary disease or disease that is metastatic at the time of diagnosis. These include antiestrogen agents such as tamoxifen and antiangiogenic agents such as bevacizumab, used as adjuvants and normally combined with at least one cycle of a cytotoxic agent. The anti-RANKL drug denosumab was also recently approved for both the prevention and treatment of metastases, but is limited to the context of bone. There are also a number of drugs in clinical trials as adjuvant therapy for recurrent cancer. Several of these involve repurposing of approved drugs such as cytotoxic agents, new-targeted immune stimulators, tumor-centric kinase inhibitors and newer antiangiogenic agents such as cediranib. Table 3 lists examples of these drugs that are being repurposed for treatment of recurrence with a summary of recent clinical findings. A few of the more recent clinical trials include new agents such as those targeting MAPK/MEK and PI3K/Akt signaling pathways, and PARP are summarized in Table 4. A commonality of the outcomes with all these agents is that while generally well tolerated, only a small number (~30%) of patients had an objective response to the drugs, if they responded at all. It is also interesting to note that none of these agents were designed to specifically target dormant micrometastatic cancer cells. Only a few agents such as tamoxifen are specifically intended to keep disseminated cancer cells dormant, and tamoxifen has only a modest effect on recurrence [Davies et al. 2013].
Repurposing of FDA-approved drugs for tumor recurrence.

Drugs in clinical trials for tumor recurrence.
Dormancy-associated genes and pathways
Genes have been identified that in model systems at least seem to prevent micrometastasis, but whether these are physiologically relevant or simply peculiarities of the models is currently less than clear. Nonetheless, these studies are beginning to identify the mechanistic underpinnings of dormancy and its escape, and as such research clarifies the situation as to which models replicate human cancer, we can expect that effective drugs will be designed to target these pathways. A recent review by Aguirre-Ghiso has provided a comprehensive overview of mechanisms associated with tumor dormancy including potential targets and therapies [Sosa et al. 2014]. The concept of targeting disseminated, dormant cancer cells and identifying potential molecular targets does have some history. For instance, Welch and his group identified KISS1 as a protein that maintains tumor dormancy and suggests that micrometastatic cells could be a legitimate target [Nash et al. 2007]. Research is beginning to identify multiple mechanisms for dormancy and escape. For example, stress signaling through the p38(SAPK) pathway and ER-stress signaling may coordinate the induction of growth arrest and drug resistance [Ranganathan et al. 2006; El Touny et al. 2013], but these are likely downstream signaling events from more proximal dormancy target(s) such as BMP7, that is secreted by the bone marrow stroma [Kobayashi et al. 2011]. More recently, epigenetic reprogramming by such agents as HDAC inhibitors has been shown to induce dormancy-like growth arrest and present a potential therapeutic strategy [Landreville et al. 2012]. Using microarray, RNAseq and deep sequencing technologies, molecular signatures of dormancy have been found, first in cancer cell lines [Kim et al. 2012], and even more recently in clinical specimens [Ross et al. 2015; Wang et al. 2015]. Without clear mechanistic targets of dormancy and given that current drug design proceeds from first identifying a target and then designing a drug to fit that target, the prospects for finding antimetastatic drugs by targeting dormancy are not likely to be realized in the near future. Are there other approaches that can be used?
Case study: development of compounds targeting dormant cancer cells using a suppressive extracellular matrix screen
Screening
Our logic was that because cancer cells are generally more resistant to conventional anticancer drugs when the cells are grown on a matrix of any kind [Sutherland et al. 1979; Vescio et al. 1987; Teicher et al. 1990; Hurst et al. 2013], that a screen to identify compounds targeting disseminated cancer cells put into dormancy by a suppressive normal ECM should show the opposite pattern. The screening method used is illustrated in Figure 1 and is described in more detail in a recent publication [Hurst et al. 2015]. Human bladder (J82) and breast cancer (MDA-MB-435) cells were plated into microplates onto plastic as actively growing monolayers or onto the suppressive ECM SISgel. SISgel is prepared from small intestine submucosa (SIS) by partial digestion with pepsin [Voytik-Harbin, 2001; Hurst et al. 2013]. The wells grown as monolayers received fewer cells than the same cells grown on SISgel to account for slower replication on the SISgel and to ensure that the monolayers had not overgrown at the time of treatment. The cells were allowed to assume their respective phenotypes, exposed to drug-like diversity chemical libraries followed 48 hours later by a cell-viability assay. Hits were defined as compounds for which the cell viability in the SISgel well was 50% or less of that in the actively growing monolayer well at the same dose, that is, the opposite of most drug screening, as illustrated in Figure 1. Conventional anticancer drugs usually yield the opposite pattern, as we have previously shown [Hurst et al. 2005]. Hits were further delineated by obtaining full dose–response relations on cells growing on SISgel and plastic and further tested on other cancer types in addition to bladder and breast. In screening a 3000-compound diversity set from the NCI, 2 hits survived the screen above. These compounds both showed limited toxicity in mice, with MTDs of 55–75 mg/kg, three times weekly. A screen of a second library of 12,000 drug-like compounds from Chembridge identified 2 additional compounds with the requisite activity in culture, but these proved too toxic in vivo to be considered further. A detailed description of the compound libraries, the hit compounds and the screening procedure is found in our original publication [Hurst et al. 2015]. Table 5 demonstrates that the differential activity observed between cells grown on plastic and on SISgel is not simply an artifact of placing the cells on SISgel because the cells behave very similarly on Matrigel, which is a ‘cancer-friendly’ matrix. Table 6 compares the activity of DT320 on SISgel and in monolayer culture for several cell lines and against the efficacy of several conventional agents.
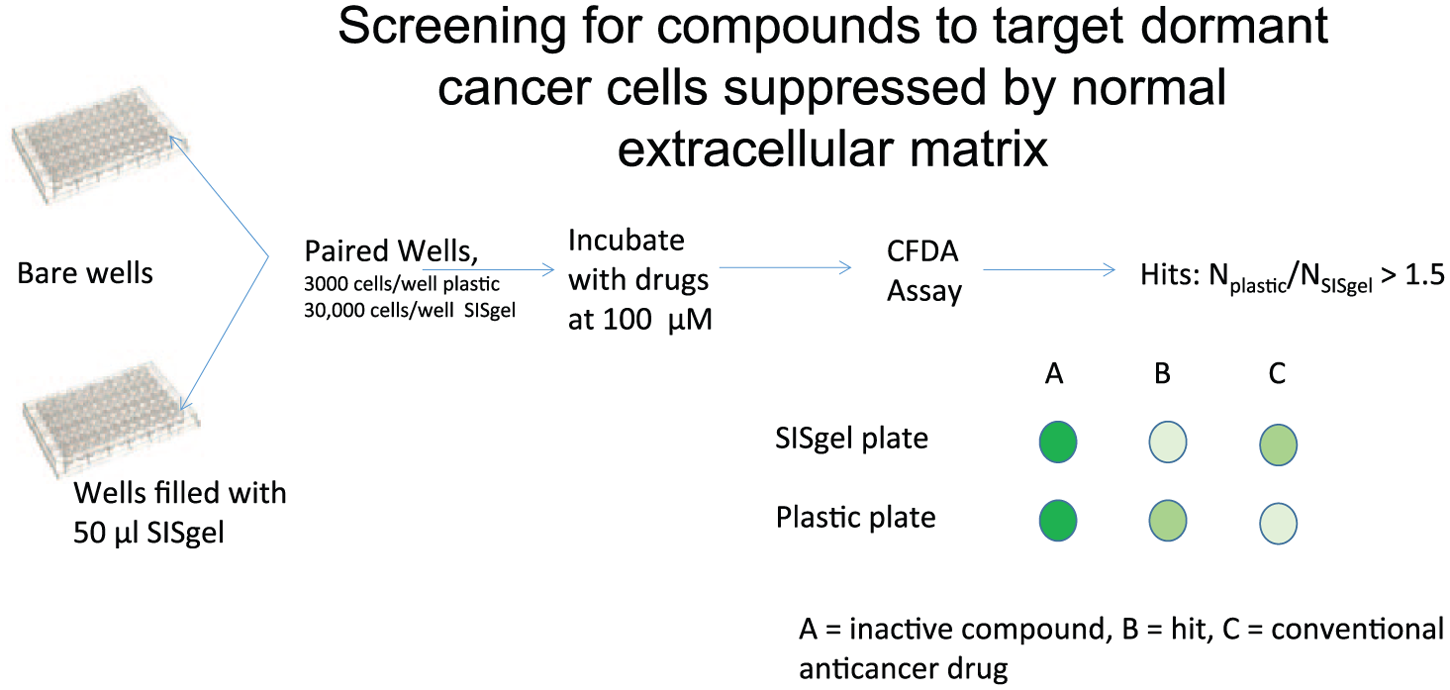
Schema of screening procedure to identify compounds that target micrometastatic cells suppressed by the normal extracellular matrix.
Comparison of EC50 values (µM) of cell kill of human breast, prostate, bladder and pancreatic cancer cell lines versus conventional agents. Cells were exposed to drugs, and the CFDA-AM assay used as a marker of cell proliferation. Data represent n = 6–8 from three separate experiments.

D, doxorubicin; G, gemcitabine; C, cisplatin.
Comparison of EC50 (µM) values of DT320 for different human cancer cell lines grown on Matrigel (fully malignant phenotype) versus SISgel (suppressed phenotype).

NS, not significant.
Effect on cancer stem cells in vitro
Because the so called ‘cancer stem cell’ is purported to be the origin of many cancers and can recapitulate tumors, we assessed the effect of our drugs on cancer stem cells, as is described in detail [Hurst et al. 2015; Mitra et al. 2015]. Briefly, breast cancer cells (4T1 cells) were sorted to obtain an aldehyde dehydrogenase 1 (ALDH1), CD44v3 high phenotype and expanded in spheroid culture. To test the efficacy of drugs required a different approach because if the cancer stem cells were plated onto plastic, they would immediately differentiate. Accordingly, cells were disaggregated and cultured again in serum-free medium, treated with DT310 or DT320 (DT) agents, and the number of metabolically active cells was assessed. The stem cell preparation yielded an approximate nine-fold enrichment of stem-cell markers. Enriched stem cells were highly resistant to doxorubicin while the DT agents had similar sensitivity for stem cells and parental cells. Significantly, breast cancer stem cells were much more sensitive to the DT agents, particularly to DT320, when compared with conventional chemotherapeutic drugs [Hurst et al. 2015].
Efficacy in vivo
Efficacy in suppressed-flank xenograft model
The drugs identified above were tested in two animal models. The first was a flank xenograft in which the fluorescently labeled breast tumor cells (MDA-MB-435 GFP) are coinjected with SISgel into an immunodeficient mouse [Hurst et al. 2013]. The SISgel then suppresses the replication of cells and substantially normalizes them so that histopathologically they resemble dysplasia, even after the SISgel has been absorbed [Hurst et al. 2013]. After 3 weeks of treatment (week 4 of experiment) the spots of suppressed cancer cells had vanished in 6/8 xenografts, whereas no response was evident in either the gemcitabine-treated (6 of 6) or untreated xenografts (8 of 8) [Hurst et al. 2015]. In the untreated animals, some of the xenografts showed evidence of escape from suppression in the form of increased intensity of the GFP label and resumption of malignant growth. The difference in response (6/8) versus gemcitabine (0/6) was significant by the Fisher’s Exact Test at p = 0.0097.
Efficacy in vivo in a natural metastasis model
Although our drugs appeared to be active in vivo in the suppressed-flank xenograft model above, this model is still somewhat artificial in that it involves SISgel. Efficacy was also tested in a physiological model of metastasis using a syngeneic triple-negative breast cancer (4T1) model in an immunocompetent mouse that we modified to slow the rate of growth and metastasis and increase its predictability [Bailey-Downs et al. 2014]. Drugs were given starting two weeks after cell implantation, to allow for micrometastatic cells to settle in the lungs without the formation of macrometastases [Bailey-Downs et al. 2014], such that the treatment is directed at preventing activation of dormant cells. The results are summarized in Table 7. All treatments had a significant effect on the number of individual micrometastatic cells, but this possibly reflected the slower growth of the primary tumors that was induced by all agents. However, the main effect of our drugs was to significantly decrease the number of large clusters and vascularized metastases [Hurst et al. 2015]. Thus the approach appears to have been successful in demonstrating the feasibility of targeting micrometastases at the stage where they begin replication using a suppressive ECM as a screening tool to identify new drugs.
Average number of clusters of cells or vascularized metastases at week 5 for 4T1 mouse syngeneic breast model of metastasis.

The p values listed are in comparison with doxorubicin. All differences in comparison with untreated animals were significant at p < 0.01 except macrometastases with doxorubicin (p < 0.05). The effect of all agents versus untreated on micrometastases was significant at p < 0.05.
Clinical perspectives
All the evidence points to the importance of targeting cancer progression by eliminating the disseminated cancer cells. Such drugs should prove most useful with patients who are disease free following definitive therapy by surgery or radiation. The fraction of patients who fit into this category varies by cancer type, and complete and accurate data are not readily available, but it represents a significant portion of all patients. A few examples are illustrative. A SEER–Medicare database search of breast cancer identified 10,798 cases of which 1833 showed a delayed recurrence (17%) [Stokes et al. 2008]. For prostate cancer patients following prostatectomy, recurrence at 5 and 10 years respectively was 13.6 and 19.9% [Xia et al. 2014]. For colon cancer, 33.4% of patients of all stages experienced recurrences after 5 years with as much as 24.7% of Stage I patients experiencing recurrence [Yang et al. 2013]. These are all lives that could be potentially saved by targeting disseminated cancer cells. Even some patients with metastasis at the time of diagnosis could potentially profit if, for example, the metastatic burden was low enough to target metastases individually (e.g. those in the liver).
In summary, the lead compounds we identified demonstrate the feasibility of targeting the dormant micrometastatic cancer cells and could light the way to identifying targets and understanding mechanisms of action. Given the slow pace of the more conventional approach of seeking to understand mechanisms of dormancy as discussed above, this approach might be more rapid.
Footnotes
Acknowledgements
The authors gratefully acknowledge the technical assistance of Brian Disch, Taleah Farasyn, Paul Hauser and Jessica Thorpe with the papers on which this review is based.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported, in part, by grants from the NIH, R01 DK069808 (REH), 1R43 CA139804, 1R01 CA136944, 1R43 CA135867 and 2R44 CA135867 (MAI), 1R43 CA168105, and from the Oklahoma Center for the Advancement of Science AR10.1-031(REH), Oklahoma Center for the Advancement of Science and Technology (OCAST), Oklahoma Applied Research Support (OARS) grant AR11-073 (MAI), and the Stephenson Cancer Center of the Oklahoma University Health Sciences Center (REH). Funding agencies had no role in the design, collection, analysis or interpretation of data.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.