
Abstract
Colorectal cancer (CRC) persists as one of the most prevalent and deadly tumor types in both men and women worldwide. This is in spite of widespread, effective measures of preventive screening, and also major advances in treatment options. Despite advances in cytotoxic and targeted therapy, resistance to chemotherapy remains one of the greatest challenges in long-term management of incurable metastatic disease and eventually contributes to death as tumors accumulate means of evading treatment. We performed a comprehensive literature search on the data available through PubMed, Medline, Scopus, and the ASCO Annual Symposium abstracts through June 2015 for the purpose of this review. We discuss the current state of knowledge of clinically relevant mechanisms of resistance to cytotoxic and targeted therapies now in use for the treatment of CRC.
Introduction
Colorectal cancer (CRC) remains a significant cause of morbidity and mortality worldwide with high disease incidence and, in spite of large-scale screening efforts recommended for all US adults, significant numbers of patients presenting with advanced, metastatic disease [Siegel et al. 2014b]. Metastatic disease is considered incurable, with the exception of patients presenting with oligometastatic lesions confined to the liver or lung amenable to resection, or metastasectomy [Abdalla et al. 2004; Adam et al. 2004]. When treatment with curative intent is not possible, patients are typically given a combination of cytotoxic chemotherapy often in conjunction with a targeted therapy. In spite of advances in systemic therapy, the 5-year survival rate is still a mere 12.5% [Siegel et al. 2014a], and the primary reason for treatment failure is believed to be acquired resistance to therapy which occurs in 90% of patients with metastatic cancer [Longley and Johnston, 2005]. Resistance to targeted therapy noted by disease progression is often noted within 3–12 months on epidermal growth factor receptor (EGFR) antagonists [Cunningham et al. 2004; Van Cutsem et al. 2007], necessitating a change in treatment.
Malignant tumors can have intrinsic resistance and/or acquired resistance and each is important in determining initial and subsequent lines of treatment. Innate resistance is typically noted during early drug development or in early phase clinical trials of biologic efficacy, however sometimes innate resistance is not understood until retrospective analyses of in vivo studies. For instance, resistance to EGFR antagonists was not well understood and from initial studies only 10–20% of patients exhibited a response to the EGFR targeted therapies cetuximab or panitumumab [Cunningham et al. 2004; Bardelli and Siena, 2010]. The subsequent elucidation of RAS mutations in CRC clarified a marker of innate resistance to these therapies, and changed their clinical use.
Different mechanisms of acquired resistance can exist for each cytotoxic therapy and each targeted pathway, but often acquired resistance to one drug confers resistance to other drugs which may work by different mechanisms of action, a concept referred to as multidrug resistance (MDR). In general, resistance to traditional cytotoxic therapy is accomplished by decreasing the delivery of drug to the cancer cell, either by increased efflux out of the cell mediated by ATP-dependent transporters, by decreased uptake into the cell, or by a change in enzymes involved in metabolism. Alternately, resistance can be conferred by changes within the cell itself by genetic or epigenetic modifications that can alter drug sensitivity [Gottesman et al. 2002].
Resistance to targeted therapies occurs by different mechanisms including upregulation, mutation, or activation of downstream signaling molecules within specific pathways; pathway bypass mechanisms; or increased cross-talk between analogous pathways [Longley and Johnston, 2005; Tejpar et al. 2012]. Understanding the mechanisms of acquired drug resistance to targeted therapies is critical for the development of novel, rational, and more effective treatment combinations and will help guide future therapies.
Since the development of markedly improved genomic sequencing and molecular biology techniques, a plethora of new putative mechanisms of resistance and potential therapies have been realized. We will review some of these mechanisms and their clinical relevance.
Cytotoxic/cytostatic chemotherapy
Cytotoxic chemotherapy has long been the backbone of treatment for CRC in patients with lymph node positive and metastatic disease. Efficacy was first recognized in the US Intergroup INT-0035 trial in 1990 which showed the beneficial effect of 5-fluorouracil (5-FU) over surgery alone [Moertel et al. 1990]. Since the adoption of widespread use of chemotherapy for advanced CRC in the adjuvant and metastatic setting, other cytotoxic drugs have been studied and validated including capecitabine, tegafur, irinotecan, and oxaliplatin, leading to their incorporation into practice guidelines and widespread use in clinical practice. The details of this pathway described below are depicted in Figure 1.

Schematic representation of the fluoropyrimidine pathway.
Fluoropyrimidines (FPs) such as 5-FU and capecitabine exert antitumor activity by inducing a state of thymidylate deficiency and creating imbalances in the nucleotide pool, which leads to impaired DNA replication, transcription, and repair and subsequent cell death [Wilson et al. 2014]. 5-FU was one of the first chemotherapeutic drugs reported to have anticancer activity [Heidelberger et al. 1957]. Following the earliest publications noting its efficacy in colon cancer, 5-FU became the mainstay of therapy and is still given as a standard part of most treatments for advanced or metastatic CRC (mCRC).
One inherent mechanism of resistance to FPs in a subset of tumors is microsatellite instability (MSI), or replication error (RER+). Microsatellites are repetitive genetic sequences, typically 1–5 base pairs repeated 15–30 times, and instability in these regions due to either insertion or deletion of repeated units causes alteration in the DNA replication process. Sporadic mutations in mismatch repair (MMR) genes leading to MSI occur in 10–20% of CRCs [Carethers et al. 2004]. Mutated MMR genes, such as MLH1, MSH2, MSH6, PMS1, or PMS6 and loss of their respective proteins, can be caused by germline mutations as seen in nearly all cases of hereditary nonpolyposis colon cancer (Lynch syndrome), or by sporadic mutations [Wheeler et al. 2000]. The loss of MMR proteins causes diffuse errors in repetitive DNA sequences, resulting from loss of scanning and recognizing errors during DNA replication and failure to edit these errors to maintain an intact genetic code [Fink et al. 1998]. The loss of these proteins is typically a result of epigenetic hypermethylation of the promoter regions of both alleles of the MLH1 genes, which halts gene expression [Wheeler et al. 2000]. Deficiency in the subsequent protein products of the hMSH2, hMLH1 and hMSH6 MMR genes and loss of detection of mismatched and unpaired bases is thought to be a primary mechanism of inherent resistance to FPs because cells become tolerant to DNA damage and do not undergo apoptosis [Carethers et al. 2004]. In one study, MMR deficient cell lines that had MMR function restored by inserting a corrected clone of hMLH1 gene led to increased sensitivity to FPs [Meyers et al. 2001].
5-FU
5-FU (Adrucil; Pfizer, Teva Pharmaceuticals) is the prototypical FP, a synthetic fluorinated pyrimidine analog, administered intravenously. It works by multiple mechanisms to create fluorinated nucleotides that are incorporated into DNA in place of thymidine (dTMP, or deoxythymidine monophosphate) thus inhibiting DNA replication and causing cell death. It is a prodrug that requires intracellular conversion to its active metabolites. The primary active metabolite is FdUMP (fluorodeoxyuridine monophosphate) which inhibits the enzyme thymidylate synthase (TS), a key enzyme in the creation of the DNA nucleotide dTMP. Inhibition of TS prevents conversion of dUMP to dTMP, which in turn impairs DNA synthesis in the S phase of the cellular replication cycle. In addition, other 5-FU metabolites 5-fluorouridine triphosphate (5-FUTP) and 5-fluorodeoxy triphosphate (5-FdUTP) are created by alternate enzymatic pathways and create false nucleotides which are incorporated into DNA and interfere with normal protein production leading to cell death [Nicum et al. 2000].
One of the most well established mechanisms of resistance to 5-FU, and other antifolates, is increased expression of TS, the primary target of the metabolite FdUMP. TS expression is a key predictor of 5-FU activity [Longley et al. 2003], and its expression has long been recognized as a primary determinant of resistance [Berger et al. 1985]. This is supported by studies showing an inverse association between low tumor TS levels and improved response rates to 5-FU [Salonga et al. 2000; Johnston et al. 1995; Leichman et al. 1995]. TS is encoded by the TYMS gene, and genetic alterations occur by copy number variations or by variations in the TYMS promoter region [Muhale et al. 2011]. In one study, patients with CRC with low TYMS gene expression had improved median survival compared with those with higher TYMS expression [Leichman et al. 1997]. This concept was further validated by at least two published meta-analyses showing an inverse relationship of TYMS gene expression with survival and response to FP therapy [Popat et al. 2004; Qiu et al. 2008].
Increased expression of TS is also an acquired mechanism of resistance after exposure to 5-FU therapy [Chu et al. 1993]. Normally, unbound TS (not bound by the metabolite dUMP) binds to its own mRNA in a negative feedback loop to inhibit its own translation, in turn reducing levels of the enzyme. It is hypothesized that TS bound by FdUMP cannot enact this negative feedback, resulting in increased protein expression and decreased sensitivity to 5-FU [Longley et al. 2002].
The inhibition of TS by the metabolite FdUMP requires formation of a complex between FdUMP and 5, 10-methylenetetrahydrofolate (CH2THF). Normally, multiple enzymes regulate the intracellular level of folate in order to maintain an appropriate pool of tetrahydrofolate, which is formed by the reduction of CH2THF to 5-methyltetrahydrofolate (CH3THF) by the enzyme methylenetetrahydrofolate reductase (MTHFR). MTHFR activates a unidirectional reaction to convert CH2THF to CH3THF, thereby decreasing the amount of CH2THF and in turn decreasing the activity of TS. Decreased enzymatic activity of MTHFR increases the concentration of the reduced cofactor CH2THF and increases the inhibition of TS by increased concentration of the FdUMP-CH2THF complex. Several single nucleotide polymorphisms (SNPs) have been shown to affect the activity of MTHFR. Two in particular are the 677C>T and 1298A>C polymorphisms. These decrease the activity of the enzyme and were observed in vivo to show mixed results, with one study demonstrating a lack of independent correlation to response [Marcuello et al. 2006] but numerous others showing a greater response to 5-FU based therapy [Sohn et al. 2004; Etienne-Grimaldi et al. 2010]. Although it seems intuitive that increased activity of this enzyme would lead to resistance, studies have not shown that upregulation or amplification of the MTHFR gene or protein product correlate with chemoresistance.
Fluorouracil must go through several enzymatic steps before it is converted to its other active metabolites 5-FdUTP, and 5-FUTP. It has been shown that activity of three of the necessary enzymes for these conversions, thymidine phosphorylase (TP), uridine phosphorylase (UP), and orotate phosphoribosyl transferase (OPRT), correlate with the sensitivity of CRC cells to the cytotoxic effects of 5-FU [Schwartz et al. 1985; Houghton and Houghton, 1983].
TP is an enzyme encoded by the TYMP gene and is responsible for converting 5-FU to 5-fluoro-2-deoxyuridine (5-FUDR), an intermediate in the conversion of 5-FU to the active metabolite 5-FdUMP. It has been observed that expression of TP correlates with response to 5-FU therapy [Panczyk, 2014]. Cells with higher levels of TP theoretically should have greater sensitivity to 5-FU due to increase in the concentration of FdUMP. To date, however, studies have shown mixed results regarding response to 5-FU-based chemotherapy and level of TP expression. Low TP expression, measured in one study by reverse transcriptase polymerase chain reaction (RT-PCR) and in another by immunohistochemistry (IHC) and tissue microarrays, correlated with improved treatment outcomes, in terms of overall survival (OS), in patients with mCRC treated with adjuvant 5-FU [Soong et al. 2008; Salonga et al. 2000]. A more recent study analyzed tumor tissue from mCRC by RT-PCR and found longer time to progression with high TP expression [Lindskog et al. 2014]. Further well-designed trials are needed to establish a definitive link between TP expression and resistance to FP therapy.
OPRT is the protein product of the uridine monophosphate synthase (UPMS) gene. This enzyme catalyzes the conversion of 5-FU to 5-fluoro-uridine monophosphate (5-FUMP), an intermediate but necessary step in the production of the active metabolites 5-FUTP and 5-FdUTP. Increased expression of UPMS gene and increased levels of OPRT in tumor tissue has been shown in multiple trials to increase the chemo-sensitivity of cells to 5-FU [Koopman et al. 2009; Tokunaga et al. 2007; Isshi et al. 2002]. Only one study, however, has demonstrated that decreased expression, by means of UPMS knockdown cell lines, leads to resistance to 5-FU [Muhale et al. 2011]. More research is needed to determine if this is a clinically significant biomarker of sensitivity or mechanism of resistance in vivo.
Dihydropyrimidine dehydrogenase (DPD) is the enzyme primarily responsible for catabolism of 5-FU to 5-fluorodihydrouracil (5-FUH2), also referred to as dihydrofluorouracil (DHFU), the first step in 5-FU elimination. 5-FUH2 is then converted into the soluble molecule 5-fluoro-β-alanine and eliminated in urine. Intrinsic overexpression of DPD by malignant cells has been shown in vitro to extend resistance to 5-FU [Longley and Johnston, 2005; Takebe et al. 2001]. High levels of DPD mRNA expression in CRC cells have also been associated with 5-FU resistance [Salonga et al. 2000]. This has been demonstrated as an intrinsic mechanism of resistance, but available data is not yet conclusive on DPD as a means of acquired resistance. In addition, DPYD gene expression has been investigated as a biomarker of treatment resistance, but this has also not been shown to be clinically relevant [Yanagisawa et al. 2007; Vallbohmer et al. 2006].
Numerous variable number of tandem repeats (VNTRs) and single nucleotide polymorphisms (SNPs) of the TYMS, MTHFR, DPYD, and UPMS genes have been identified as contributors to 5-FU resistance, and could serve as potential targets for future directed therapy to combat drug resistance, or potentially for gene therapy [Panczyk, 2014].
Oral FPs
Capecitabine, S-1, and tegafur-uracil are three oral FPs demonstrating similar efficacy as intravenous 5-FU, with the potential for more convenience by reducing time spent in an infusion suite or on an infusion pump. Capecitabine is the only one of these currently approved for use in the United States. All of these therapies are prodrugs which, by a series of enzymatic reactions, are eventually converted to 5-FU in the tumor microenvironment. Because of this, they have many of the same mechanisms of resistance as 5-FU, however with additional layers needed for activation come new potential sources of resistance.
Capecitabine
Capecitabine (Xeloda; Genentech, Roche, Switzerland) is an oral FP carbamate which was developed to mimic continuous 5-FU infusion, but with activation occurring primarily at the tumor site. After absorption it is converted to fluorouracil by three enzymes, two of which, TP and UP, are present in higher concentrations within malignant cells compared to normal cells [Wilson et al. 2014]. This orally administered drug was designed to function similar to infusional 5-FU, giving a consistent level of drug exposure to tumor cells over time. Since the final steps in the pathway occur preferentially in the tumor it has a theoretical advantage of increased efficacy and decreased systemic toxicity, however this has not been the case in clinical trials [Cassidy et al. 2011].
The efficacy of capecitabine in advanced CRC, including stage III and metastatic disease, was originally demonstrated by two phase III randomized trials completed in the mid-1990s comparing capecitabine to weekly bolus 5-FU/LV [Van Cutsem et al. 2001; Hoff et al. 2001]. These results have been subsequently confirmed in a meta-analysis of six randomized trials that showed equivalent OS in patients treated with single agent and matched capecitabine-containing regimens to single agent and matched 5-FU-containing regimens [Cassidy et al. 2011].
Since capecitabine is eventually converted into 5-FU within the tumor, many of the mechanisms of resistance are identical to those implicated for 5-FU. Several of these mechanisms have been evaluated specifically in capecitabine treated cells. As shown in relation to 5-FU, DPD expression has been implicated in resistance to capecitabine. In one study, higher gene expression, as measured by mRNA analysis from tumor tissues following capecitabine treatment, correlated with resistance to therapy as evidenced by shorter progression-free survival (PFS) and lower response rate in patients [Vallbohmer et al. 2007]. In a retrospective review of 556 tumors from the CAIRO (CApecitabine, IRinotecan, Oxaliplatin) study, DPD expression inversely correlated with PFS and OS [Koopman et al. 2009]. This study also investigated the predictive value of multiple other markers including OPRT, TP, TS, and ERCC1, and found that high OPRT in stromal cells was favorable for response to therapy, but elevated expression in tumor cells correlated with worse PFS and OS. The reason for this is unclear and has not been fully elucidated.
TP converts 5-FU prodrugs into active metabolites that exert antitumor effect. This enzyme is expressed in higher concentration in tumors, but was shown to be expressed in the tumor microenvironment rather than the tumor cells. TP has other actions related to carcinogenesis, including promotion of metastasis, tumor infiltration, and angiogenesis which correlate with poor prognosis [Ye and Zhang, 2013]. High expression of TP, however, correlates with better response to capecitabine, and loss of function confers resistance [Petrioli et al. 2010], a fact that has also been shown to have clinical significance with improved response to CAPIRI chemotherapy by extending time to progression [Meropol et al. 2006]. This is seen in contrast to the inverse effect of TP expression on response to 5-FU. One putative mechanism of loss of function of TP is abnormal pre-mRNA splicing by increased levels heterogeneous nuclear RNP (hnRNP) H/F splicing factors which leads to loss of function of the TYMP gene [Stark et al. 2011]. Further research is ongoing in regards to exact mechanisms of development of resistance specifically to capecitabine that may be distinct from other FPs.
S-1 and tegafur-uracil
S-1 (TS-1; Taiho Pharmaceutical, Tokyo, Japan) is a fourth generation oral FP available worldwide outside of the United States. It consists of tegafur (UFT), gimeracil (5-chloro-2, 4-dihydroxypyridine) and oteracil (potassium oxonate). Tegafur is a prodrug that is converted to fluorouracil within tumor cells. Gimeracil is an inhibitor of DPD, the primary enzyme responsible for fluorouracil metabolism. Oteracil inhibits the phosphorylation of fluorouracil in the gastrointestinal tract and serves to reduce toxic side effects of 5-FU [Sakuramoto et al. 2007]. Tegafur–uracil is another oral FP agent and is approved in 50 countries worldwide. It consists of tegafur attached to uracil, which blocks DPD degradation of fluorouracil’s pyrimidine base. Tegafur is not well tolerated by patients as they experience consistently high toxicity negating any benefit, which is the primary reason for lack of approval in the United States.
The approval of S-1 is based on two trials in East Asian patients. It is considered an acceptable alternative to 5-FU when combined with oxaliplatin as part of FOLFOX or XELOX [Hong et al. 2012], or an alternative to FOLFIRI when combined with irinotecan [Muro et al. 2010] in East Asian patients. A subsequent meta-analysis of eight trials from Japan and China in Asian patients with either mCRC or advanced gastric cancer demonstrated essentially equivalent efficacy to the 5-FU-containing regimens [Cao et al. 2014].
Tegafur is metabolized by the cytochrome P-450 isoenzyme encoded by the CYP2A6 gene. The expression of the variants CYP2A6*4 and CYP2A6*1B seem to be the primary determinants for the degree of conversion into 5-FU, and are required for the anticancer activity of tegafur [Wang et al. 2011]. This has been shown to correlate with toxicity, a putative reason why white patients experience intolerable toxicity compared with East Asian patients [Shirao et al. 2004]. It has been shown that patients with wild-type CYP2A6 have improved efficacy outcomes compared with mutants in gastric cancer, and also showed increased response to TIROX compared with several polymorphisms [Kim et al. 2013], presumably related to decreased conversion to 5-FU and exposure of tumor cells.
TAS-102
TAS-102 (Taiho Pharmaceutical, Tokyo, Japan) is an oral nucleoside antitumor agent with multiple components. Trifluorothymidine (TFT) is the active ingredient and is a FP that is active in inhibiting DNA replication. TFT is phosphorylated by thymidine kinase (TK) and in turn inhibits thymidine synthase (TS) in a similar fashion to other FPs. It can also form the triphosphate form, TFT-TP, which is incorporated into DNA thus interfering directly with replication [Wilson et al. 2014]. In a phase II trial comparing monotherapy with placebo, TAS-102 showed improvement in both PFS and OS in a small subset of patients with mCRC refractory to traditional therapy [Yoshino et al. 2012]. The results of the phase III RECOURSE trial comparing TAS-102 to placebo in pretreated patients showed improvement in OS and PFS in European patients with mCRC refractory to at least two prior lines of therapy [Mayer et al. 2015]. As yet there is no direct experimental evidence supporting a process for development of resistance, however one would presume it may follow a pattern very similar to other FPs.
Numerous gene polymorphisms of OPRT, MTHFR, UGT1A1, and DPD have been investigated in relation to toxicity [Tsunoda et al. 2011; Choi et al. 2012], but to date none of these have demonstrated clinical significance in relation to chemoresistance. These oral FPs may prove useful for patients in the United States if they gain FDA approval.
Irinotecan
Irinotecan (Camptosar, formerly CPT-11; Pfizer, Pharmacia) is a semisynthetic analog of the natural alkaloid camptothecin, a DNA topoisomerase I inhibitor. This enzyme relaxes super-coiled double-stranded DNA by inducing single strand breaks. By inhibiting its action, irinotecan interferes with DNA replication and transcription. Topoisomerase I inhibitors stabilize intermediate cleavage complexes formed between the inhibitor, the enzyme, and the DNA single strand and subsequently prevent DNA re-ligation, leading to cell death. Irinotecan must be converted to its active metabolite SN-38 to exert its anticancer effect [Xu and Villalona-Calero, 2002], and SN-38 reversibly binds to topoisomerase-1 and stabilizes the complex. It was approved in an accelerated fashion by the FDA in 1996 and received full approval for use in CRC in 1998.
The addition of irinotecan to adjuvant therapy for advanced CRC and to mCRC was supported by multiple trials starting in the early 1990s [Douillard et al. 2000; Saltz et al. 2000b; Giacchetti et al. 2000]. Single-agent irinotecan has only a modest response rate of 11–27% in clinical trials in 5-FU refractory patients with mCRC [Cunningham et al. 1998], but a larger effect when given in combination with FPs, 5-FU and capecitabine, and/or oxaliplatin [Shimada et al. 1996]. In the first-line setting response rates range from 39% to 49% [Saltz et al. 2000a; Douillard et al. 2000]. It has been approved as first-line therapy in advanced or metastatic disease, but not for adjuvant treatment as this was shown to provide no additional benefit to FP monotherapy [Van Cutsem et al. 2009]. The addition of irinotecan to FP-based regimens also shows a significant benefit in PFS from 4.3 to 7 months, and OS from 12 to 18–21 months, and its efficacy as monotherapy in patients with FP refractory disease indicates activity in spite of acquired resistance to FP therapy [Cunningham et al. 1998; Rougier et al. 1998].
Irinotecan resistance in CRC appears to develop by a few mechanisms including low intratumor level of the active metabolite SN-38, a decrease in expression of topoisomerase I, change in the activity of the SN-38-Topo I- DNA complex, and changes in downstream events such as suppression of apoptosis, cell cycle alterations, or enhancement of DNA repair.
The level of intratumoral SN-38 can be altered by increased efflux or increased metabolism of either irinotecan or the metabolite SN-38. Active transport out of cells by the multidrug resistance protein (MRP), an ATP-binding cassette (ABC) transporter protein, has been shown in cancer cells to result in resistance to irinotecan and SN-38 [Longley and Johnston, 2005; Thomas and Coley, 2003]. Numerous in vitro studies of polymorphisms of transporter proteins, such as ABCC1/MRP1, ABCC2/MRP2, and ABCG2/BCRP have shown results that explain variation in drug toxicity in patients, and the development of drug resistance against irinotecan and SN-38 [Zhao et al. 2014]. However, these results have been inconsistent in the literature and there are no in vivo studies to support development of these proteins as targets for therapy.
The active metabolite SN-38 is created by hydrolysis of CPT-11 by carboxylesterases CES1 and CES2. Carboxylesterase activity in cancer cells has been shown to correlate with sensitivity to irinotecan in in vitro studies [Kojima et al. 1998; Boyer et al. 2004], however, outside of associations with specific SNPs with cytotoxicity, no direct correlation has been shown between carboxylesterase activity or expression and chemoresistance. SN-38 is metabolized by glucuronidation in the liver by the enzyme uridine diphosphate glucuronosyltransferase (UGT) to form SN-38 glucuronide (SN-38G). Increased glucuronidation-mediated clearance in CRC cells may contribute to tumor resistance to irinotecan [Cummings et al. 2002]. Genetic variants of both liver and plasma UGT1A, by VNTRs and other polymorphisms, have been shown to have effects on CPT-11 toxicity including neutropenia and diarrhea, however these have not been shown to correlate with response to therapy [Marcuello et al. 2004]. Any association with poor response to treatment is believed to be a result of having to reduce the dose of the drug due to side effects, not from the polymorphism [Carlini et al. 2005; Rouits et al. 2004].
Irinotecan itself is metabolized by oxidation, primarily by two hepatic cytochrome P-450 enzymes, CYP3A4 and CYP3A5. These enzymes metabolize irinotecan to APC ((7-ethyl-10-(4-N-aminopentanoic acid)-1-piperidino) carbonyloxycamptothecin) and NPC (7-ethyl-10-(4-amino-1-piperidino)carbonyloxycamptothecin), respectively. NPC is an inactive metabolite but can be hydrolyzed by hepatic carboxylesterase back to SN-38 [Panczyk, 2014]. Variations in CYP3A4 activity have been investigated as a source for resistance to treatment with irinotecan, however current research, including studies measuring in vivo activity of these enzymes, is inconclusive about the role of numerous described polymorphisms [Xie et al. 2004; Fujiwara and Minami, 2010].
As described previously, SN-38 binds to topoisomerase I as it binds to DNA to relieve strand tension during replication. The action of irinotecan is dependent on normal functioning topoisomerase I, encoded by the TOP1 gene. Level of TOP1 gene expression and copy number was demonstrated as a potential cause of intrinsic resistance by an in vitro study of colon cancer cell lines [McLeod and Keith, 1996]. A more recent Scandinavian retrospective analysis of tumor tissue from irinotecan pre-treated patients identified an increased objective response in tumors with increased copy number of TOP1 gene, although this did not reach statistical significance [Nygard et al. 2014]. Increased copy number has been noted in as many as two-thirds of tumors from a cohort of patients with stage III CRC [Smith et al. 2013], so low levels of TOP1 gene copy number is implicated as a means of intrinsic resistance.
SN-38 forms non-covalent, but stable bonds to the Topo-1-DNA complex to exert its cytostatic effect that leads to cell death. Changes in the binding site of topoisomerase I prevent SN-38 from creating a stable bond. One study of irinotecan-treated tumor samples identified that point mutations in the Top1 gene, as detected by RT-PCR of mRNA from tumor samples, altered the binding of SN-38 to the enzyme, implicating acquired resistance to therapy [Tsurutani et al. 2002]. Further in vitro analyses of SN-38 resistant tumor cell clones showed that resistance may develop by decreased affinity of TOP1/SN-38 binding or by mutations in the linker domain which lead to decreased flexibility of the complex [Gongora et al. 2011].
Again, numerous SNPs have been discovered to help explain the genetics behind these mechanisms of resistance, as well as explain potential variations amongst individuals with apparently similar tumor types [Panczyk, 2014]. The clinical relevance of these numerous polymorphisms is not yet certain based on the current body of literature.
Oxaliplatin
Oxaliplatin (Eloxatin; Sanofi-Aventis Pharmaceuticals) is a third generation platinum compound that works by inducing DNA cross-linkages leading to apoptotic cell death. It is a square planar platinum, distinct from other platinums in that it contains a bidentate ligand 1,2-diaminocyclohexane in lieu of two monodentate ammine ligands. It causes inter- and intra-strand DNA cross-links that halt replication and transcription [Hector et al. 2001]. It was approved for use in Europe in 1996 and it was granted accelerated approval in 2002 by the US FDA, with full approval granted in 2004 for use in combination with 5-FU for advanced CRC or mCRC.
Oxaliplatin has been shown to have minimal single agent activity, with response rates of around 20–24%, in several phase II trials [Becouarn et al. 1998; Diaz-Rubio et al. 1998] in the front-line setting. In combination with 5-FU and leucovorin (FOLFOX), however, it has been shown in multiple trials to contribute to increased PFS and OS compared with 5-FU and leucovorin alone with response rates as high 50% [de Gramont et al. 2000]. The GERCOR study published in 2004 [Tournigand et al. 2004] showed equivalence of the two primary regimens FOLFOX and FOLFIRI, which was subsequently confirmed by Colucci and colleagues the following year [Colucci et al. 2005]. Since the FOLFIRI regimen was shown to be ineffective in the adjuvant setting, FOLFOX has become the mainstay of therapy in postoperative patients.
The mechanisms of resistance of oxaliplatin appear to be somewhat different from those seen with cisplatin and carboplatin. In fact, it has been shown to be active in cancer cell lines resistant to earlier generation platinum compounds [Raymond et al. 2002]. It has also been well established that enhanced replicative bypass and loss of MMR are mechanisms of resistance to cisplatin but not to oxaliplatin by both in vitro and in vivo studies. One study comparing similar platinum resistant cell lines exposed to cisplatin and oxaliplatin showed that oxaliplatin exerted cytotoxic effects at a much lower concentration than did cisplatin, and also created fewer DNA-Pt adducts [Hector et al. 2001]. These adducts are likely different than those created by cisplatin, supported by the fact that MMR deficient cells demonstrate resistance to cisplatin but not oxaliplatin [Fink et al. 1997], implying that even MMR deficient cells are able to recognize oxaliplatin DNA-Pt adducts and undergo apoptosis.
Like other chemotherapy, resistance can occur by decreased entrance into or increased efflux out of the tumor cells. The transporters regulating platinum efflux are ABC-type MDR proteins as well as copper-transporting p-type ATPases [Panczyk, 2014]. A few transporters have been recognized to regulate influx of platinum compounds, including copper transporter (CTR) proteins, organic cation transporters (OCTs), and an undefined cis-specific platinum influx transporter that has not been implicated in oxaliplatin resistance. Some transporters have been investigated, including members of the SCL22 family (such as OCT2), as well as CTRs [Zhang et al. 2006] (including CTR1 and CTR2 [Holzer et al. 2006]), and these have been shown in preclinical studies to be potential targets of modulation of influx and efflux. No in vivo studies have shown actual clinical significance of these postulated resistance mechanisms. In vitro studies have shown that oxaliplatin requires lower concentrations to exert anticancer effects, but these transports have not been shown to be major mechanism of resistance.
A mechanism of platinum compound inactivation is the formation of conjugates, or covalent linkages, between the drug and the thiol glutathione (GSH, a tripeptide of glutamic acid, cysteine, and glycine) [Meijer et al. 1992]. Glutathione is a potent antioxidant that functions to prevent oxidative damage to DNA and RNA. This also serves as a mechanism of increased clearance, however, because GSH–platinum conjugates become a substrate for the ABC transporter proteins which promotes drug efflux out of the cell [Ishikawa and Ali-Osman, 1993]. It has been shown that some tumors may be resistant to platinums due to their having higher levels of GSH [Kelland, 1993]. There are also studies demonstrating the potential influence of several polymorphisms of the glutathione s-transferases, however this is not consistently shown and has not been demonstrated in in vivo studies [Panczyk, 2014]. A meta-analysis of five clinical studies showed no correlation of a common polymorphism, 313A>G, with response to oxaliplatin-based therapy [Ye et al. 2013].
Platinum compounds exert their antitumor effect by creating adducts in the DNA, typically intra-strand cross-links [Hector et al. 2001], that lead to apoptosis once recognized by MMR mechanisms. Nucleotide excision repair (NER) is a mechanism by which cells repair DNA damage and one of the ways in which cancer cells overcome chemotherapy effects. This may be particularly true for platinums by removing DNA-Pt adducts from the strand. Excision repair cross-complementation group 1 and 2 (ERCC1 and 2) proteins are two of the main effectors of the NER mechanism. They recognize DNA-Pt adducts and coordinate the base excision process. High expression of ERCC1 mRNA has been associated with poor response to FOLFOX in 5-FU resistant tumors [Shirota et al. 2001], implicating that enhanced DNA repair decreases the benefit of platinum therapy. Oxaliplatin-resistant tumors have upregulation of ERCC1 as shown by higher mRNA expression in retrospective analysis of tumor samples [Baba et al. 2012] implicating that this may also be a form of acquired resistance. Numerous polymorphisms in the ERCC1 gene have been identified and investigated as predictive, however most have shown inconclusive results [Panczyk, 2014]. The ERCC1 (354C>A) SNP, in conjunction with the XRCC1 (1196A>G) SNP have an independent predictive effect on disease control rate and OS [Liang et al. 2010]. This, however, must still be taken in the context of a retrospective analysis and we await further prospective analyses.
Epigenetic changes have also been implicated in development of resistance, primarily hypermethylation. Although oxaliplatin works by inducing single-strand breaks by intra-strand adduct formation, it also forms inter-strand adducts and can cause double-stranded breaks. These abnormalities are repaired by the NER process, as well as by BRCA1, which works by homologous recombination to repair double-strand breaks [Fedier et al. 2003]. Inactivation of the BRCA1 interactor SRBC by hypermethylation of the SRBC1 gene is associated with oxaliplatin resistance [Moutinho et al. 2014]. Little has been definitively discovered about the impact of epigenetic alterations on CRC resistance, however, as in other tumor types, this is likely a mechanism.
Resistance to cytotoxic chemotherapy occurs by several variations on similar themes, such as decreased intracellular drug concentration, altered metabolism, or alterations to targets of the therapy. These mechanisms of resistance are summarized in Table 1. Some of these concepts translate into mechanisms of resistance to novel targeted therapies, however often resistance is more complex, at times not involving mutation but molecular pathway alteration, something not seen with evasion of traditional chemotherapy.
Reported mechanisms of resistance to chemotherapy agents.

Reported mechanisms of resistance to targeted therapies.

BMDC, bone-marrow-derived cell; EGFR, epidermal growth factor receptor; HGF, hepatocyte growth factor; IL, interleukin; KRAS, Kristen rat sarcoma; NRAS, neuronal rat sarcoma; PTEN, phosphatase and tensin homolog; TGF-α, transforming growth factor alpha; VEGF, vascular endothelial growth factor.
Targeted therapy
As knowledge about cancer biology and genetics expands, new treatment targets have been discovered and drugs developed to affect tumors in more elegant and rational fashion than impacting all cells actively in the cell cycle. This has led to treatments with good response and often less-toxic side-effect profiles than cytotoxic/cytostatic therapy. Two broad categories of targeted therapies include monoclonal antibodies and small molecule inhibitors, different in their target of action and mode of administration.
Although targeted agents provide hope for more effective therapy, studies have shown modest benefit and, like more traditional chemotherapy, are subject to both primary and secondary resistance which ultimately leads to treatment failure. Protein tyrosine kinases, such as EGFR and vascular endothelial growth factor receptor (VEGFR) are some of the most well-understood pathways of potential therapy used to treat CRC. Resistance is often seen through constitutive pathway activation, perhaps by receptor overexpression or mutation. By identifying specific ligands or receptors involved in cell growth or survival, both the activating ligand and receptor can potentially be targeted. Common mechanisms of resistance are related to alterations of the target itself, bypass mechanisms, upregulation and activation of downstream effectors, or cross-talk between associated pathways which activate complementary cell survival and growth pathways. The details of these pathways described below are depicted in Figure 2.
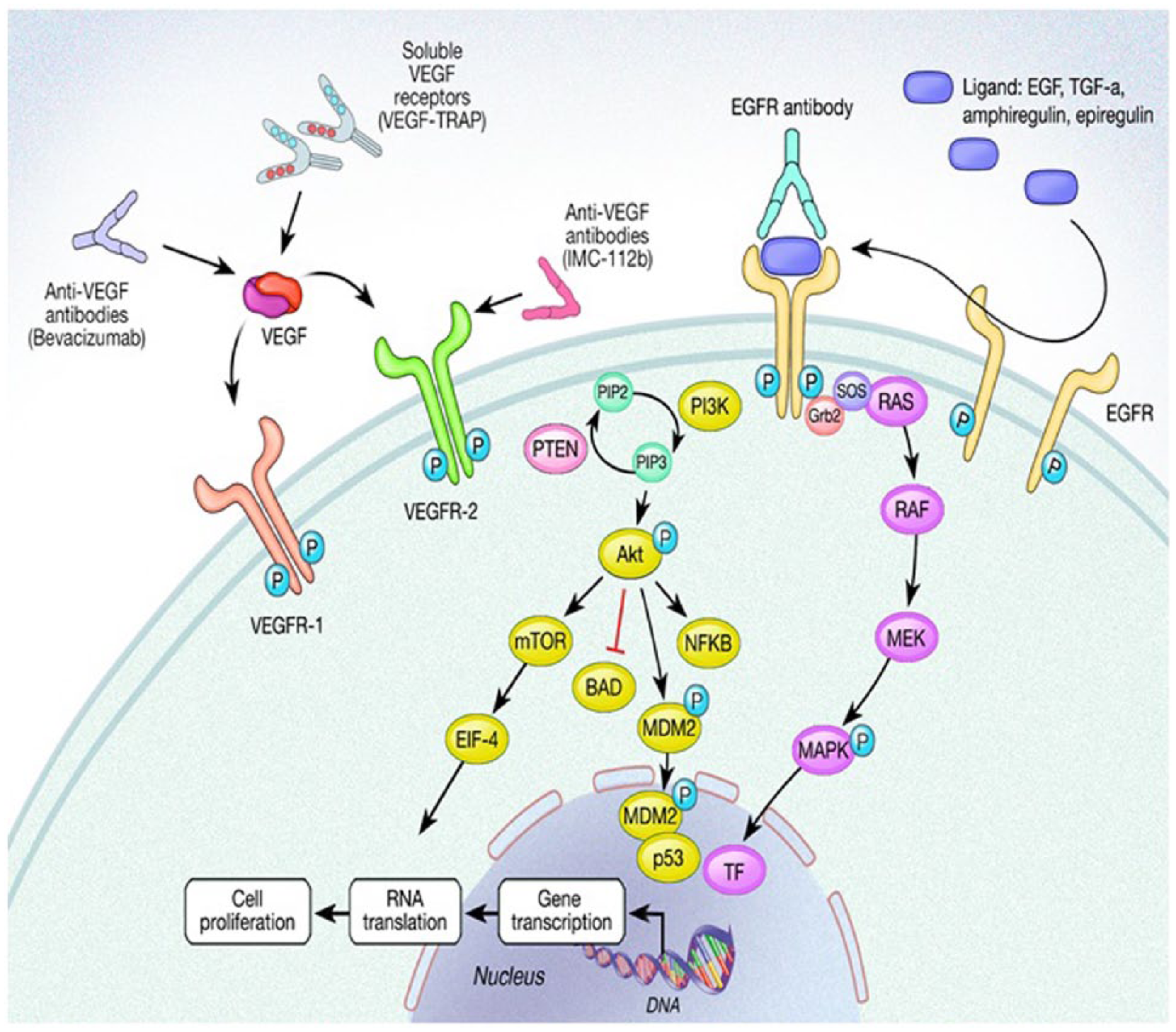
Schematic representation of the epidermal growth factor receptor (EGFR) and vascular endothelial growth factor (VEGF) and receptor (VEGF-R) pathways.
Epidermal growth factor
EGFR is a transmembrane signaling protein on human epidermal cells involved in cellular signaling for proliferation and survival, and its over-expression is linked to cancer development and proliferation. It is composed of an intracellular tyrosine kinase domain, a transmembrane hydrophobic segment, and an extracellular receptor to which ligands bind for activation. It is a member of the ErbB tyrosine kinase receptor (TKR) family, and is also referred to as ErbB1, or HER1. This family also includes human epidermal growth factor receptor 2 (HER2) which is implicated in other cancer types, also known as ErbB2. Other members of the ErbB family include Erb3 and 4. All four ErbB family members can interact with each other to induce signaling depending on which ligand is bound. EGFR is activated primarily by the binding of epidermal growth factor (EGF) or transforming growth factor-alpha (TGF-α) [Roskoski, 2014].
Inactive EGFR exists as a monomer, however once a ligand binds to the extracellular domain, this induces either homodimerization with another EGFR monomer or heterodimerization with a different ErbB family receptor. This then induces transphosphorylation of several intracellular tyrosine kinase (TK) domains which propagate downstream signaling by several well-described pathways, most notably the Ras/Raf/MEK/ERK1/2/MAPK and PI3K/Akt/mTOR pathways [Roskoski, 2014; Spano et al. 2005]. The PI3K/Akt pathway has been shown to participate primarily in mediating cell survival and motility invasion, where the Ras/Raf/MEK/ERK1/2 pathway is implicated in cellular proliferation. Both of these pathways are also involved in angiogenesis, cellular adhesion, cell motility, development, and organogenesis [Roskoski, 2012].
Initiation of the Ras/Raf/MEK/ERK pathway by EGFR begins with activation of Grb2 (growth factor receptor bound protein 2) adaptor protein which binds to the phosphotyrosine residue of the ErbB receptor as well as SOS (son of sevenless), a Ras-guanine nucleotide exchange factor [Lowenstein et al. 1992]. SOS activates Ras to Ras-GTP which propagates downstream signaling, starting with Raf and resulting in cell growth and differentiation [Roskoski, 2012].
Activation of the PI3K tyrosine kinase by EGFR induces phosphorylation of membrane-bound phosphatidylinositol 4,5-bisphosphonate (PIP2) to form phosphatidylinositol 3,4,5-triphosphate (PIP3). PIP3 attracts Akt (also known as protein kinase b, or PKB) to the plasma membrane [Longley and Johnston, 2005]. Akt is a serine/threonine kinase that binds tightly to PIP3 and interacts with various other kinases, including phosphoinositide-dependent protein kinase 1 (PDK1) and mammalian target of rapamycin complex 2 (mTORC2). These kinases both phosphorylate Akt and stimulate it to phosphorylate and activate mTOR, another serine/threonine kinase which has multiple targets involved in cell survival. Activated Akt also phosphorylates, and thus inactivates, Bcl2 associate death promotor (BAD), which, in its unphosphorylated state, is pro-apoptotic by binding and sequestering the antiapoptotic Bcl2. Akt also has the ability to affect apoptosis by phosphorylating MDM-2 which, once translocated to the nucleus, downregulates p53 expression [Feng et al. 2004]. Thus, by activation of the PI3K/Akt pathway, EGFR promotes cell survival via both mTOR and Bcl2 activation and by p53 downregulation [Roskoski, 2014].
Phosphatase and tensin homolog (PTEN) is a negative regulator of the PIP3KCA/Akt pathway, and is one of the most frequently mutated tumor suppressor genes [Yamada and Araki, 2001]. Its normal function is to regulate the activity of PIP3 by dephosphorylation, and thus inhibit Akt activity. Mutation of PTEN and loss of its activity thus leads to constitutive activation of Akt and promotion of cell survival.
Upregulation or activation of EGFR is present in 60–80% of CRCs [Messa et al. 1998; Porebska et al. 2000; Salomon et al. 1995] and overexpression of EGFR is associated with poor prognosis in mCRC [Mayer et al. 1993]. As such, this receptor has been a focus of drug development and targeted therapies against this receptor are approved for use in CRC, including cetuximab and panitumumab.
Cetuximab
Cetuximab (Erbitux; Bristol-Meyer Squibb, Eli Lilly and Company) is a chimeric mouse/human IgG1 monoclonal antibody that binds to the extracellular domain of the EGFR. Cetuximab was first approved by the FDA for use in mCRC in February 2004 as monotherapy or in combination with irinotecan. Cetuximab was shown to have a response rate of about 10% as monotherapy in a study of irinotecan pretreated patients by Cunningham and colleagues with improved time to progression from 1.5 to 4.1 months [Cunningham et al. 2004]. In addition, Jonker and colleagues showed an 8% partial response rate and improved PFS, as well as improved overall quality of life compared with best supportive care in heavily pretreated patients with mCRC [Jonker et al. 2007]. Interestingly, the addition of cetuximab to irinotecan in irinotecan-resistant/refractory disease showed prolongation of both PFS and OS compared with cetuximab monotherapy, suggesting a means of overcoming resistance to irinotecan [Cunningham et al. 2004].
The current understanding of the mechanism of action of cetuximab is that the drug binds to the external domain of the EGFR and prevents ligand binding, preventing cell growth and survival. After binding, the receptor is internalized and degraded without activation or phosphorylation [Tabernero, 2007]. There is also evidence that cetuximab-receptor binding also induces antibody-mediated cytotoxicity, leading to tumor cell death [Ciardiello and Tortora, 2008]. In addition, cetuximab was shown to down-regulate VEGF expression, thus reducing tumor angiogenesis [Ciardiello et al. 2000].
Early in the investigation into mechanisms of resistance, it was postulated that somatic EGFR mutations may play a significant role in lack of response, as was demonstrated to other targeted therapies, for example in lung and breast cancer [Paez et al. 2004; Hudis, 2007]. It has since been suggested by large cohort studies that EGFR mutations are not only uncommon in mCRC, but that they do not impact response when they are present [Barber et al. 2004; Moroni et al. 2005]. However, one specific point mutation of EGFR, a change of serine to arginine at codon 492 (S492R) leading to a change in the external domain of the EGFR was recently shown to confer resistance to cetuximab binding but not the other approved EGFR targeted therapy panitumumab [Van Emburgh et al. 2014]. It has been shown that this mutation is likely present only in patients following exposure to EGFR antagonism, and not in treatment-naïve patients [Esposito et al. 2013]. Recently, the incidence of this mutation in cetuximab and panitumumab treated tumors was evaluated in a review of tumor samples from the phase III ASPECCT trial of second-line treatment of wild-type KRAS exon 2 mCRC [Price et al. 2014]. Of the 999 patients, roughly half in each group had post-treatment EGFR S492R status available, and the mutation was found in 1% of panitumumab treated tumors and 16% of cetuximab treated tumors. The mutation was not identified in any of the pretreatment samples, confirming that this is an acquired mutation conferring resistance. In addition, these EGFR mutant tumors demonstrated longer duration of treatment before progression, but had lower median OS by almost 2 months [Price et al. 2015].
Although the relationship between EGFR mutations and response remains unclear, there is evidence that increased copy number of the EGFR receptor, as measured by fluorescent in situ hybridization (FISH), is a positive predictor of response to treatment with both cetuximab and panitumumab [Sartore-Bianchi et al. 2007; Cappuzzo et al. 2008]. Likewise, low-level expression is purported as a means of intrinsic resistance to therapy, yet this has not been shown clinically.
In early clinical trials, only about 10–20% of unselected patients with mCRC responded to single agent EGFR antagonists. It is now known that tumors with a mutation of the Kristen rat sarcoma (KRAS) gene found on chromosome 12, encoding a small G-protein downstream of EGFR, have inherent resistance to both cetuximab and panitumumab, leading to minimal clinical effect [De Stefano and Carlomagno, 2014]. Lievre and colleagues first noted that tumor response was dictated by KRAS mutation status when they examined 30 tumors and found that 13/30 tumors harbored a KRAS mutation and two thirds (68%) of the nonresponders had a mutation whereas this was found in none of the responders [Lievre et al. 2006]. The first large retrospective review of treated tumors found that 43.2% of tumors had at least one mutation in exon 2 which correlated with lack of response to cetuximab [Karapetis et al. 2008]. The predictive mutations were noted first in exon 2, codons 12 and 13, however subsequent studies have shown that as many as 5–11% of additional tumors have mutations in exons 3 (codons 59 and 61) and 4 (codons 117 and 146) which are similarly predictive of lack of response to EGFR antagonist therapy [Therkildsen et al. 2014; Misale et al. 2014].
The discovery of activating KRAS mutations led to better patient selection, however, in spite of ‘wild type’ EGFR, still only about 40% of patients responded to EGFR targeted therapy, indicating an alternate means of innate resistance. KRAS amplification has been identified as a cause of resistance to anti-EGFR therapy as well, but is likely present in only in ~1–2% of cases and it is mutually exclusive from KRAS mutations [Misale et al. 2014]. Resistance to EGFR antagonists is also influenced by neuronal RAS (NRAS) mutations [Douillard et al. 2013; Meriggi et al. 2014].
NRAS is a gene found on the short arm of chromosome 1 which encodes for a GTPase membrane enzyme that shuttles between the Golgi and the cell membrane [De Stefano and Carlomagno, 2014]. Mutations have been identified at exon 2 (codons 12 and 13), exon 3 (codons 59 and 61) and exon 4 (codons 117 and 146) and occur at a frequency of approximately 2–5% among patients with mCRC. These mutations have been shown to negatively impact response to anti-EGFR therapy, with either cetuximab or panitumumab [Meriggi et al. 2014; De Roock et al. 2010; Therkildsen et al. 2014]. Mutations in NRAS are mutually exclusive of mutations in KRAS and BRAF. In spite of increasing knowledge, there are still tumors that do not respond to treatment, indicating some yet unidentified innate or intrinsic resistance. For now, it is standard of care to test not only for KRAS mutations in exons 2 [Allegra et al. 2009], 3 and 4, but also NRAS mutations prior to exposing a patient to anti-EGFR therapy.
Activating KRAS mutations have been shown to be a mechanism of both intrinsic and acquired resistance. In a large analysis by Misale and colleagues, EGFR sensitive cell lines of primary colon tumors and metastases showed that numerous molecular alterations, mostly point mutations in KRAS gene, led to both cetuximab and panitumumab resistance. This study also showed that KRAS mutations could be detected in blood samples as early as 10 months before radiographic evidence of disease progression, implicating that this type of resistance may occur quite early in disease treatment [Misale et al. 2012].
Changes in pathways related to the target of therapy, or parallel to it, may confer resistance. In regard to EGFR targeted therapies, PIK3CA activating mutations are seen in 14.5% of untreated tumors, most in exons 9 and 20 [De Roock et al. 2010]. These mutations have been shown to predict lack of response to EGFR therapy in pre-clinical models and early in vivo studies [Jhawer et al. 2008; Sartore-Bianchi et al. 2009; Sood et al. 2012]. The largest analysis to date, however, with more than 750 untreated CRC tumor samples from European centers showed an association between exon 20 and worse response to cetuximab therapy, but not exon 9, perhaps implicating limitations of tumor sample volume in prior studies that showed association with exon 9 [De Roock et al. 2010]. Additionally effecting the EGFR/PI3K/Akt pathway is epigenetic inactivation of PTEN phosphatase which has been suggested to be predictive of lack of response to EGFR targeted therapy [Perrone et al. 2009]. Loss of function in PTEN has been observed to occur by means of mutation, loss of gene expression, or hypermethylation of the promotor region [Sood et al. 2012]. Studies have shown that patients with tumors having preservation of PTEN and wild-type PIK3CA had improved OS and a trend toward improved PFS [Sood et al. 2012], and that time to progression was significantly shorter in tumors with PIK3CA mutations and loss of PTEN [Saridaki et al. 2011]. In a retrospective analysis of cetuximab treated patients, none of the patients with loss of function PTEN mutation showed response to cetuximab therapy [Frattini et al. 2007]. In addition, BRAF mutations, present in about in 9.6% of CRC [Hirschi et al. 2014], have been shown to confer a worse prognosis, and may contribute to EGFR therapy resistance, however this has not been definitively demonstrated in clinical trials.
Many of the aforementioned mutations and alterations play a central role in innate, or primary resistance. It has been shown that approximately 50% of tumors harbor a KRAS mutation at time of progression on treatment with anti-EGFR therapies [Hobor et al. 2014; Misale et al. 2012], implying that other means of resistance subvert the effect of EGFR blockade. Heterogeneity within a given tumor is often observed where mutated cells (e.g. KRAS, NRAS, BRAF mutated) exist along with other sensitive, non-mutated cells, implicating a potential protective mechanism. In addition, it is known that EGFR has multiple ligands other than EGF, including TGF-α and amphiregulin, which bind to the receptor and activate downstream signaling. A paracrine method of resistance has been postulated in which tumor cells can increase the secretion of these alternate ligands which serve to protect the cells from EGFR-blockade. This was shown in an in vitro study of cetuximab-resistant cells cultured with cetuximab-sensitive cells where the sensitive cells grew in the presence of cetuximab and increased secretion of ligands and increased EGFR signaling were observed [Hobor et al. 2014].
Lastly, another mechanism of acquired resistance may occur by alteration of complementary pathways which serve to bypass the EGFR antagonism, so-called escape mechanisms. In particular, upregulation of ErbB2 (HER2) and MET can occur and may contribute to resistance. ErbB2 is a receptor with no known biologic ligand that undergoes a conformational change that activates the PI3K/Akt pathway [Rajput et al. 2007]. Amplification of HER2 has been noted in only 2–3% of untreated tumors, however has been shown that cetuximab-resistant cell lines demonstrate both HER2 amplification as well as an increase in secretion of heregulin, a ligand which binds to the HER2 receptor [Yonesaka et al. 2011; Ciardiello and Normanno, 2011; Bertotti et al. 2011]. MET gene amplification may act as another escape mechanism, as well as increased binding by its ligand hepatocyte growth factor (HGF) [Bardelli et al. 2013]. The vascular endothelial growth factor (VEGF) pathway, described in detail later in this review, is also closely related to the EGFR pathway. In fact, overexpression of VEGF is one way in which tumor cells overcome resistance to EGFR inhibition. It has been shown that EGFR over-expression leads to upregulation and increased signaling by VEGF, however the resistance seems to occur independent of EGFR signaling [Tabernero, 2007]. This was shown in a study of gene expression of biomarkers from tumors pretreated with irinotecan or oxaliplatin, followed by single agent cetuximab. Those tumors with elevated VEGF expression were more resistant to cetuximab [Vallbohmer et al. 2005].
Panitumumab
The other approved EGFR monoclonal antibody is panitumumab (Vectibix; Amgen), a fully human IgG2 monoclonal antibody that binds with high affinity to the extracellular domain of the EGFR. It is thought that panitumumab prevents downstream activation signaling by competitive inhibition [Hohla et al. 2014]. It was approved by the FDA for use in mCRC expressing EGFR in September 2006 in combination with other approved chemotherapy regimens for patients who had progressed on or after initial therapy. It has also now been approved, as of May 2014, for use in the first-line setting with FOLFOX therapy.
The efficacy of panitumumab was first demonstrated by Van Cutsem and colleagues as monotherapy [Van Cutsem et al. 2007]. In the ASPECCT trial, a phase III randomized noninferiority trial, panitumumab was shown to have similar OS and PFS as cetuximab, showing similar efficacy in patients with exon 2 KRAS wild-type mCRC [Price et al. 2014]. Perhaps the largest and most impactful study of this agent has been the PRIME trial which compared FOLFOX4 with and without panitumumab. In patients with KRAS wild-type tumors, there was a significantly improved PFS of 9.6 months with the addition of panitumumab, compared with 8 months in the control arm. Objective response rate was also improved at 55%, compared with 48% in the FOLFOX4 alone group. Again, this effect was lost in those with mutated KRAS. In fact PFS was significantly worse in this group compared with FOLFOX4 alone [Douillard et al. 2010].
Retrospective analyses of tumor samples from trials of tumors treated with panitumumab have shown that KRAS mutations in exon 2, codons 12 and 13, were mutated at the same frequency, approximately 43%, as in analogous reviews of trials with cetuximab [Amado, 2008]. Resistance to panitumumab is akin to cetuximab occurring by many of the same mechanisms, including mutations of KRAS, NRAS, PIK3CA, PTEN [Douillard et al. 2013; Sood et al. 2012]. One difference is that of a specific point mutation of the EGFR gene, S492R, which was shown to confer resistance to cetuximab post-treatment, but not panitumumab. The incidence of the S492R mutation in panitumumab-treated tumors has been reported from retrospective analysis of tumors from patients treated in the ASPECCT trial and found in only 1% of post-treatment evaluable tumor samples, implying that this may not be a prominent mechanism of acquired resistance. No other specific polymorphisms are published that show resistance unique to panitumumab [Van Emburgh et al. 2014].
VEGF
The role of angiogenesis and lymphangiogenesis in tumor growth is well established. Hypoxia in the tumor microenvironment has been shown to upregulate hypoxia inducible factor (HIF), which then signals production of VEGF [Maxwell et al. 1997]. Over-expression of VEGF gene and high levels of circulating VEGF protein are both associated with worse prognosis in CRC [Jurgensmeier et al. 2013]. Several agents have been developed to inhibit angiogenesis and thus slow tumor growth. VEGF has been identified as the prominent effector of angiogenesis, and has been the target of drug developments since it was recognized that its inhibition suppresses tumor growth [Kim et al. 1993]. The VEGF family is composed of at least nine ligands, VEGF-A through E, and placental growth factor (PlGF) 1 through 4. These ligands act upon at least one of three known receptors, VEGFR-1 through 3. Blocking members of this family, either the ligand or receptor, prevents tumor mitigation of local hypoxia and starvation. The first VEGF/VEGFR targeted drug brought to market was bevacizumab, and much of our understanding about resistance to VEGF inhibition comes from experience with this drug.
Bevacizumab
The monoclonal antibody bevacizumab (Avastin; Genentech/Roche) is a recombinant humanized IgG1 antibody against all isoforms of VEGF-A, a ligand for the VEGF receptors 1 and 2 [Tejpar et al. 2012]. It was first approved by the FDA in February 2004 for use in combination with chemotherapy in the first-line treatment of mCRC.
Bevacizumab has been shown to have clinical activity in CRC in numerous studies, the first of which was a trial by Hurwitz et al. [2004] which showed an improved response rate, PFS and OS with the addition of bevacizumab to a first-line irinotecan-based regimen (IFL) in mCRC. It has also been shown to have increased PFS in combination with oxaliplatin-based chemotherapy, combined with both 5-FU and capecitabine, as first-line therapy in mCRC [Saltz et al. 2008]. Both of these studies showed an improvement of no more than 2 months in PFS. Interestingly, it has also been shown that continuation of bevacizumab after progression, while changing the cytotoxic chemotherapy regimen, resulted in improved PFS and OS compared with post-progression chemotherapy alone [Bennouna et al. 2013]. The reasons behind this are likely related to the different mechanisms of resistance to angiogenesis inhibitors, including both evasive/acquired resistance and intrinsic indifference [Bergers and Hanahan, 2008].
VEGF inhibitors such as bevacizumab work by reducing the formation of new vasculature needed by developing tumors for continued growth. They induce a state of local hypoxia, however hypoxia induces increased VEGF expression via HIF, so its very inhibition induces its production. In addition, the binding of VEGF to its target leads to downstream upregulation of VEGF, creating a positive feedback loop of continued vascular growth promotion. CRC cells express VEGF receptor and demonstrate this autocrine signaling, which promotes cell survival [Mesange et al. 2014]. This occurs particularly under external stress, including stress from treatment with 5-FU [Samuel et al. 2011]. In vitro, bevacizumab resistant cell lines show strong autocrine HIF-VEGF-VEGFR signaling as a response to exposure to anti-VEGF therapy [Mesange et al. 2014].
Acquired resistance to anti-angiogenic agents is often referred to as evasive resistance, because unlike resistance to cytotoxic therapy or EGFR antagonists, this typically occurs by adapting to the presence of anti-angiogenic agents in lieu of more definitive mechanisms such as mutation of constituents within the pathway. This evasive resistance occurs by at least three different mechanisms including revascularization by alternate angiogenic pathways, development of protective mechanisms for vasculature, and enhanced ability of tumors to invade or metastasize.
Continued or revascularization can occur by upregulation of alternate VEGFR ligands. By recognizing elevated pro-angiogenic biomarkers in tumors following treatment with bevacizumab, alternate pro-angiogenic pathways have been discovered. In one study, PIGF, a ligand of VEGFR-1, was found to be significantly elevated in tumors following treatment with FOLFIRI plus bevacizumab [Kopetz et al. 2010]. This ligand, as well as VEGF-D, was shown in another study to be upregulated for about 6 weeks following treatment with bevacizumab plus chemotherapy, indicating this as a transient alteration [Lieu et al. 2013]. In addition, IL-8, a chemokine with many functions including angiogenesis, was shown to provide HIF-independent angiogenic stimulus in vitro in CRC cell lines [Mizukami et al. 2005]. Elevated pretreatment levels of IL-8 in blood were associated with shorter PFS in a phase II trial investigating biomarkers in relation to response to FOLFIRI plus bevacizumab [Kopetz et al. 2010]. Third, fibroblast growth factor (FGF) upregulation has been noted in pancreatic neuroendocrine [Casanovas et al. 2005] and glioblastoma [Batchelor et al. 2007] tumor cell lines, implicating this as a potential evasive mechanism of resistance to bevacizumab, although this has never been shown explicitly in CRC. Additional biomarkers of putative significance are platelet-derived growth factor-C (PDGF-C) [Crawford et al. 2009], neuropilin-1 (NRP-1) [Pan et al. 2007], and delta-like ligand-4 (Dll4) [Ridgway et al. 2006].
Another method of evasion of bevacizumab therapy is by increased protection of vasculature by recruitment of alternative pro-angiogenic factors and by increasing protective barriers over existing vasculature to increase its survival. Bone-marrow-derived cells (BMDCs), including vascular progenitors and vascular modulatory cells, are recruited to the tumor environment in the setting of hypoxia to promote new vessel growth [Mitchell, 2013]. Not only are progenitors of epithelial cells and pericytes recruited to the area, but so too are other vascular modulators such as tumor-associated macrophages [Pollard, 2004], TIE2 [De Palma et al. 2005], VEGFR-1+ hemangiocytes [Hattori et al. 2002], and CD11b+ myeloid cells [Yang et al. 2004]. These function to promote vascular growth by producing cytokines, growth factors and proteases that promote vascular growth and development [Bergers and Hanahan, 2008]. It has also been noted that tumors treated with anti-angiogenic agents have a change in the structure and concentration of pericyte coverage over existing vasculature [Kamba and McDonald, 2007], presumably lending to the observed rapid reconstitution of vasculature following removal of angiogenic blockade [Mancuso et al. 2006]. Two such factors contributing to recruitment of BMDCs and monocytes/macrophages to the tumor microenvironment are HIF1α and PlGF [Ribatti, 2008].
Some tumors treated with continuous anti-angiogenic therapy may also develop augmented invasiveness leading to increased local invasion and metastasis. The mechanism by which they are able to invade is by co-option of normal vasculature allowing this to provide nutrients necessary for continued growth. This was first seen in glioblastoma cell lines [Rubenstein et al. 2000], and has been since recognized in pancreatic neuroendocrine tumors as well [Casanovas et al. 2005]. Tumor cells are able to migrate along the blood vessels, a process called perivascular invasion, allowing for subsequent direct invasion into surrounding tissue [Du et al. 2008].
In some trials, a minority of patients had rapid progression in spite of angiogenic blockade, suggesting either rapid development of resistance or the presence of intrinsic resistance. It has been postulated that some of the same mechanisms of acquired resistance are present innately within some tumors, such as BMDCs such as CD11+ monocytes [Shojaei et al. 2007]. Some tumors may also intrinsically possess the invasive phenotype allowing them to co-opt surrounding normal vasculature to promote growth [Du et al. 2008].
Lastly, KRAS mutation status has been observed to affect response to bevacizumab-containing regimens when examined in subgroup analyses. One example is the TLM trial of bevacizumab continuation after progression on a bevacizumab containing first-line treatment which was designed with an exploratory endpoint of evaluation of OS, PFS, and subsequent anticancer treatment according to KRAS mutation status [Bennouna et al. 2013]. Patients treated with bevacizumab plus chemotherapy had improved PFS regardless of KRAS mutation status, however patients with KRAS wild-type cancer had improved OS with addition of bevacizumab to second-line chemotherapy that was not seen in the patients with KRAS mutant cancer. These data seem to imply a mechanism of resistance, however the treatment by KRAS status interaction test for this subgroup was negative for PFS and OS, indicating no identifiable KRAS mutation-independent treatment effect.
Given the specificity of bevacizumab to the VEGF-A ligand, and the range of evasive mechanisms within tumors, it is not surprising that resistance develops somewhat rapidly. Resistance is not considered permanent, and as mentioned above, ongoing treatment with bevacizumab has shown prolonged PFS and OS compared with chemotherapy alone. Perhaps the use of a different agent to attempt broader angiogenic inhibition, including some of the evasive mechanisms of resistance, would be more efficacious.
Aflibercept
Aflibercept (Zaltrap; Regeneron Pharmaceuticals, Sanofi-Aventis), referred to in the United States as ziv-aflibercept, is a recombinant decoy VEGFR1 and VEGFR2 fusion protein, each linked via the Fc segment of IgG1, that has anti-angiogenic and vascular permeability activity by targeting multiple members of the VEGF family, including VEGF-A, VEGF-B and placental growth factor 2 (PlGF-2). Binding these growth factors prevents their activity at the VEGFR-1 and VEGFR-2 receptors, which are found on the surface of endothelial cells and leukocytes. Its activity results in regression of tumor vasculature, inhibition of new vascular growth and remodeling of surviving vasculature [Mitchell, 2013]. It was approved for use in the US by the FDA in August 2012.
The efficacy of aflibercept was demonstrated in the VELOUR trial, a phase III randomized, placebo controlled trial of over 1200 patients with mCRC who had progressed on oxaliplatin-based chemotherapy. Patients were randomized to receive FOLFIRI plus aflibercept or FOLFIRI plus placebo. The addition of aflibercept resulted in improved response rate (20% versus 11%, respectively), PFS (6.9 versus 4.7 months, respectively), and modestly improved OS by 1.5 months [Van Cutsem et al. 2012]. This is to date the only published phase III trial demonstrating efficacy of aflibercept in any setting. Interestingly, subgroup analysis has shown that the benefit of adding aflibercept was seen regardless of whether the patient had received bevacizumab as part of first-line treatment, potentially implicating efficacy beyond development of resistance to VEGF inhibition [Tabernero et al. 2014].
Resistance to aflibercept is presumed to occur by many of the same mechanisms implicated in resistance to bevacizumab. It has been specifically shown that VEGF-C is upregulated in tumors treated with the VEGF-trap aflibercept, suggesting that alternate angiogenesis pathways are triggered to promote resistance [Li et al. 2014]. As aflibercept is studied in earlier treatment lines for mCRC, more data on acquired resistance may become available upon review of treated tumors.
Ramucirumab
Ramucirumab (Cyramza; Eli Lilly and Company, US) is a recombinant, fully humanized IgG1 monoclonal antibody directed against the extracellular domain of the VEFGR2. It binds to this receptor with high affinity, and thereby blocks ligand binding, primarily VEGF-A but others as well. It was approved in April 2015 for use in combination with FOLFIRI for the treatment of patients with mCRC who have progressed on or after prior therapy with bevacizumab, oxaliplatin, and a FP.
Approval was based on the results of the RAISE trial, a phase III randomized, double-blind, multicenter international trial of 1072 patients who had disease progression during or within 6 months of the last dose of first-line therapy [Tabernero et al. 2015]. The patients received FOLFIRI plus either ramucirumab or placebo, randomized in a 1:1 ratio. The primary endpoint, median OS, was improved by 1.6 months (13.3 versus 11.7 months) and the survival benefit was seen across all subgroups including KRAS mutants and those with short time to progression (<6 months) after previous first-line treatment. PFS was also prolonged by 1.2 months (5.7 versus 4.5 months), but there was no appreciable difference in response rate. The results of this trial indicate that VEGF receptor blockade after progression through alternate angiogenesis blockade provides additional benefit, similar to what was seen in the TLM trial with bevacizumab [Bennouna et al. 2013] and the VELOUR trial with aflibercept [Tabernero et al. 2014]. Since this anti-angiogenic therapy is still new in the armamentarium of colon cancer treatments, there is no published data to specifically implicate unique mechanisms of resistance.
Multi-kinase targeted agents
Regorafenib
Regorafenib (Stivarga; Bayer HealthCare Pharmaceuticals) is an oral multi-kinase tyrosine kinase inhibitor that targets multiple angiogenesis targets including VEGFR1, VEGFR2, VEGFR3, and TIE2, as well as multiple oncogenic targets including KIT, RET, RAF1, BRAF, and BRAF-V600E, and tumor microenvironment targets including PDGFRα (platelet derived growth factor receptor) and β, and FGFR (fibroblast growth factor receptor) 1 and 2, and p38 MAP kinase [Grothey et al. 2013; Tejpar et al. 2012].
It was approved by the FDA in September 2012 for use in patients with previously treated mCRC who had received all previously approved treatments including VEGF- and EGFR-active agents. Its approval was based primarily on the results of the CORRECT trial, a large international randomized, double-blind, placebo controlled phase III trial involving 760 patients who had previously been treated with chemotherapy plus bevacizumab, as well as an anti-EGFR drug if the patient’s tumor was KRAS wild type. The study achieved its primary end point of improved median OS by showing an improvement of 1.4 months with regorafenib treatment over best supportive care. The study also demonstrated an improvement in PFS, though there was no difference in overall response rate [Grothey et al. 2013].
Given the nature of this multi-targeted kinase, potential mechanisms of resistance are difficult to elucidate. Presumably, mechanisms of resistance previously shown for other specifically targeted agents may be applicable to this agent as well, however this agent simultaneously blocks multiple members of a single pathway and members of parallel pathways between which cross-talk may occur, though likely not all isoforms of each target. The modest clinical benefit over best supportive care (1.4 month improvement) implies that there are very likely some mechanisms of resistance. This benefit was seen in heavily pretreated patients, so it is likely the tumors had substantial acquired resistance to typical pathways of targeted treatment. Further research on this topic is certainly expected after its recent approval and with more patients being treated with this drug.
Conclusions
This review demonstrates that much is known about resistance to cytotoxic and targeted therapies in the treatment of CRC, though clearly more needs to be discovered. The goal of elucidating mechanisms of resistance is to develop methods to overcome the resistance to advance our fight against this disease and achieve better, more durable treatment responses and longer patient survival. With so many potential targets for therapy, personalized treatments with existing drugs would be expected to show improved results. Early trials of personalized therapy based on mutation status of KRAS, BRAF, PI3KCA, and expression of Topo-I, ERCC1, TS, and TP did not show any improvement in PFS [Cubillo et al. 2014]. However, some authors have proposed using KRAS, BRAF, PI3KCA, and PTEN mutation status as a signature to guide therapy [Bardelli and Siena, 2010; Sartore-Bianchi et al. 2009]. Other clinical trials involving personalized therapies for CRC based on more comprehensive genomic and molecular profiling are underway. Certainly, in this era of more targeted therapies and much improved capabilities in genomic and molecular profiling, much is left to discover about the effects that these targeted therapies have on the intricate web of signaling and activities both intracellularly and in the tumor microenvironment. Ultimately, new discoveries will continue to translate into improved treatment options and important clinical outcomes for patients.
Footnotes
Acknowledgements
We would like to acknowledge Margaret McKinney for material support.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The author(s) declare(s) that there is no conflict of interest.