
Abstract
For more than a decade, S-1 has been investigated aggressively against non-small cell lung cancer (NSCLC) in Japan. Recently, two randomized phase III trials of S-1 combined with cisplatin (CDDP) or carboplatin (CBDCA) compared with the standard platinum doublet chemotherapy were reported. S-1 and CDDP was noninferior to CDDP and DTX in terms of overall survival (OS) (median survival time [MST] 16.1 versus 17.1 months, respectively; hazard ratio [HR] 1.013; 96.4% confidence interval [CI] 0.837–1.227). Noninferiority of S-1 and CBDCA compared with CBDCA and paclitaxel was also confirmed for OS (MST 15.2 versus 13.3 months, respectively; HR 0.928; 99.2% CI 0.671–1.283). The noninferiority design employed an upper CI limit of HR<1.322 in the former trial and HR<1.33 in the latter. S-1 combined with CDDP or CBDCA was thought to be one of the standard platinum doublet regimens in the first-line setting for patients with advanced NSCLC in Japan. Some additional interesting phase I and II studies have been published in Japan. They include studies of S-1 as first-line chemotherapy when combined with nonplatinum agents; as second-line chemotherapy; within chemoradiotherapy for locally advanced disease; and in the postoperative adjuvant setting. This review will also describe the use of S-1 for the treatment of NSCLC in these settings.
Introduction
Many studies have been conducted since S-1 was first approved for non-small cell lung cancer (NSCLC) in July 2004 in Japan. They include studies of S-1 as first-line chemotherapy when combined with cisplatin (CDDP), carboplatin (CBDCA) or nonplatinum agents; as second-line chemotherapy; within chemoradiotherapy for locally advanced disease; and in the postoperative adjuvant setting. This review will describe the use of S-1 for the treatment of NSCLC in these settings.
Outline of S-1
S-1 (S-1; Taiho Pharmaceutical Co., Ltd., Tokyo, Japan) is an oral fluoropyrimidine agent that consists of tegafur (FT), 5-chloro-2,4-dihydroxypyridine (CDHP) and potassium oxonate (Oxo) in a molar ratio of 1:0.4:1 [Shirasaka et al. 1996a]. FT is a prodrug of fluorouracil (5-FU) [Giller et al. 1967]. CDHP is a reversible competitive inhibitor of dihydropyrimidine dehydrogenase (DPD), an enzyme involved in the degradation of 5-FU [Tatsumi et al. 1987]. Thus, the degradation of FT-derived 5-FU is efficiently inhibited by CDHP, and 5-FU remains in the plasma and tumor tissue longer and at higher levels than when low-dose 5-FU is continuously infused intravenously. This results in enhancement of the antitumor effect, as documented in animal models [Shirasaka et al. 1996b]. The major toxicities of fluoropyrimidines are diarrhea and mucositis [Vogelzang, 1984]. Oxo is a reversible competitive inhibitor of orotate phosphoribosyltransferase, a phosphoenzyme for 5-FU, and is distributed at high levels in the gastrointestinal (GI) tract after oral administration, resulting in a reduction in GI toxicity caused by 5-FU (Figure 1) [Shirasaka et al. 1993].
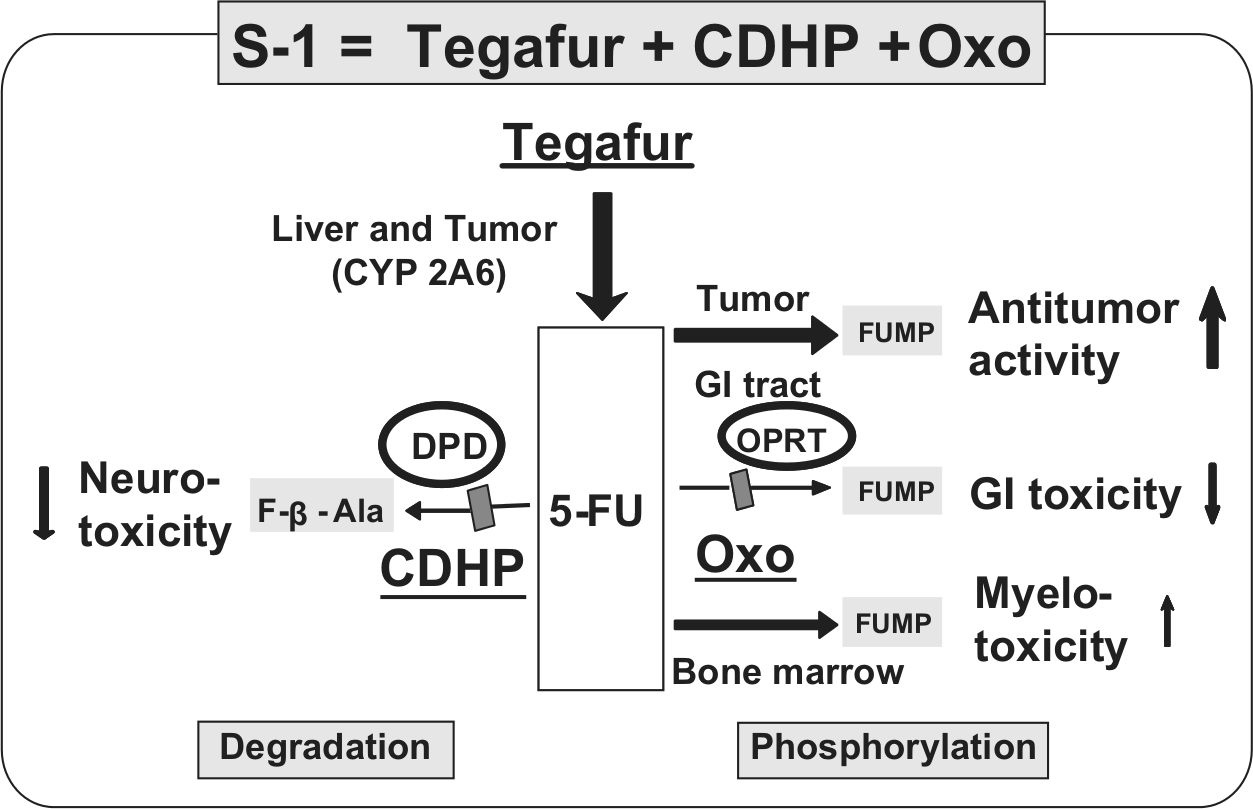
Unique metabolism of S-1. S-1 (FT, CDHP, Oxo) is an oral ‘DPD inhibitory fluoropyrimidine’ (DIF). 5-FU, fluorouracil; CDHP, 5-chloro-2,4-dihydroxypyridine; DPD, dihydropyrimidine dehydrogenase; GI, gastrointestinal; OPRT, orotate phosphoribosyltransferase; Oxo, potassium oxonate.
Early phase clinical development of S-1
S-1 in a twice daily schedule has been investigated in a phase I study in Japan. Based on the results of a phase I clinical trial, the maximum tolerated dose was 75–100 mg/body twice daily, and the dose-limiting toxicity was myelosuppression, especially leukopenia [Taguchi et al. 1997]. Note that doses of S-1 in this study were fixed and not expressed in milligrams per square meter of body surface. In an early phase II clinical trial, the initial dosing schedule was 75 mg/body twice daily for 28 consecutive days followed by a 2-week rest period. However, the dose was reduced to 50 mg/body twice daily because of skin rashes, severe myelosuppression and diarrhea. The major grade 3+ toxicities were myelosuppression and GI toxicity. When the actual administered doses were calculated according to body surface area (BSA), the rate of discontinuation of the drug because of toxicities was 71.4% at 90 mg/m2/day [Furuse et al. 2001]. Therefore, the initial dose at 80 mg/m2/day was recommended for later phase II studies. In Japan, three doses of S-1 are selected according to BSA so that they would be approximately equivalent to 80 mg/m2/day: BSA <1.25 m2, 40 mg/body twice daily; BSA 1.25 m2, but <1.5 m2, 50 mg/body twice daily; and BSA ≥1.5 m2, 60 mg/body twice daily. One cycle consisted of consecutive administration of S-1 for 28 days followed by a 14-day rest period [Hirata et al. 1999].
Different recommended doses of S-1 according to ethnicity
In contrast, the recommended doses for single-agent S-1, based on phase I studies conducted in Western countries, were 50 mg/m2 daily for 21 days every 4 weeks [Cohen et al. 2002], 40–50 mg/m2 daily for 28 days every 5 weeks [Chu et al. 2004; van Groeningen et al. 2000] or 30 mg/m2 twice daily for 28 days every 5 weeks [Hoff et al. 2003]. The toxicity profile from these phase I trials differed significantly based on the geographic region of study, with predominant hematological toxicities observed in Japanese studies and GI toxicities in studies from North America or Europe.
FT is converted to 5-FU in the liver by cytochrome P450 (CYP) enzyme. FT is hydroxylated to 5-hydroxytegafur and eventually converted to 5-FU [El Sayed and Sadee, 1982]. CYP2A6 of the CYP family is now identified as the principal enzyme responsible for the conversion process of FT to 5-FU [Ikeda et al. 2000]. However, different polymorphisms in the CYP2A6 gene among Asians and Whites, which affect its efficacy in the rate of conversion of FT to 5-FU, have been identified [Daigo et al. 2002; van der Weide and Steijns, 1999; Yoshida et al. 2003].
S-1 as first-line treatment for advanced NSCLC
A description of prospective studies evaluating first-line S-1 combined with platinum agents is given in Table 1. A phase II study of combination chemotherapy with S-1 and CDDP was conducted in previously untreated patients with advanced NSCLC, using the schedule defined by a phase I study in patients with advanced gastric cancer. This phase II study of a 3-week course of S-1 combined with 60 mg/m2 of CDDP on day 8 showed an objective response of 47%, median survival time (MST) of 11.2 months, and 1-year and 2-year survival proportions of 45% and 17%, respectively [Ichinose et al. 2004].
Prospective studies evaluating first-line S-1 combined with platinum agents.
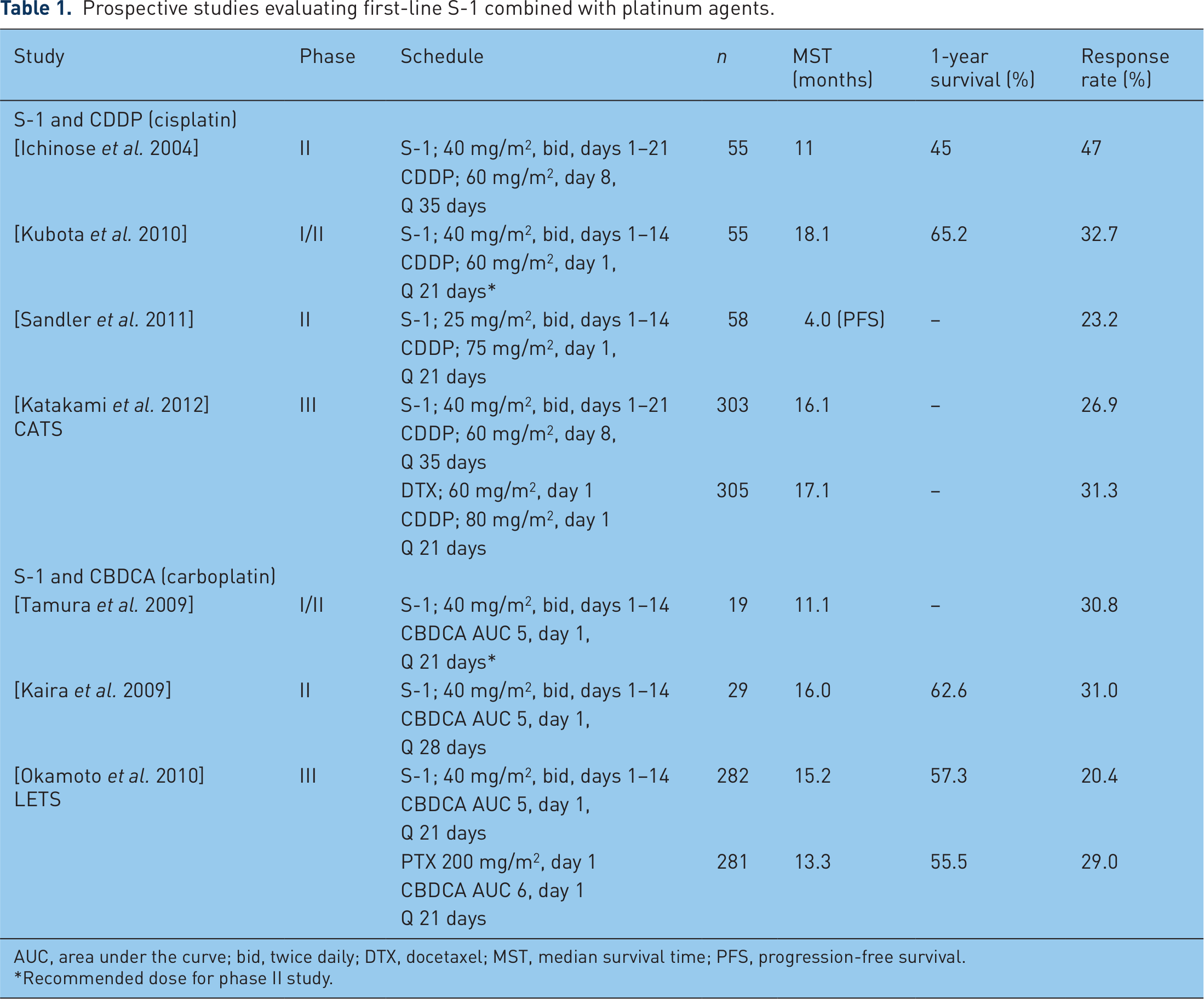
AUC, area under the curve; bid, twice daily; DTX, docetaxel; MST, median survival time; PFS, progression-free survival.
Recommended dose for phase II study.
Another phase I/II study of a 2-week course of S-1 combined with CDDP on day 1 was also conducted in Japan. Because the approved dose of CDDP for NSCLC in Japan is 70–90 mg/m2 and many regimens used day 1 administration of CDDP in a 3-week schedule, the Health, Labor and Welfare Ministry of Japan requested to evaluate the day 1 administration of S-1 and CDDP in a 3-week schedule. This phase I/II study was conducted to determine the optimal dose of CDDP on day 1 in combination with a 14-day course of S-1 within a 3-week cycle and to evaluate the efficacy and safety of the regimen at the recommended dose. In the phase I portion of the study, the maximum tolerated dose and the recommended dose of CDDP were defined as 70 and 60 mg/m2, respectively. The recommended dose of CDDP on day 1 was identical to that for the day 8 administration of CDDP used in the previous phase II study. The phase II portion of this study in 55 patients demonstrated a response rate of 32.7% (95% confidential interval [CI] 20.7–46.7%). Although the objective response did not meet the threshold level in this study, the results of survival were encouraging, with a MST of 18.1 months, a 1-year survival proportion of 65.2% and a 2-year survival proportion of 34%. The relatively good survival results might be, in part, due to second-line treatment with third-generation antineoplastic agents and/or molecular targeted agents; 74.5% of patients received second-line chemotherapy, including epidermal growth factor receptor tyrosine kinase inhibitors, as post-study treatment. In conclusion, the recommended dose of CDDP on day 1 was identical to that used for the day 8 administration of CDDP in combination with S-1 in patients with advanced NSCLC. Although this regimen did not reach the study’s primary endpoint in the later trial, the schedule of day 8 administration of S-1 and CDDP from days 1 to 21 as an experimental arm was utilized in a subsequent phase III trial [Kubota et al. 2010]. In addition, the pooled analysis from two phase II clinical studies of S-1 and CDDP in patients with previously untreated advanced NSCLC was performed. According to histological type, median progression-free survival (PFS) was 3.8 months in patients with squamous cell carcinoma (SCC) and 4.4 months in those with non-SCC. Unlike molecular targeted agents and pemetrexed (PEM), a combination of S-1 and CDDP may produce no difference in response according to histological type [Yamamoto et al. 2010b].
A randomized phase III trial, the S-1 and CDDP (CATS) study, was performed to assess whether treatment with S-1 and CDDP was as effective as CDDP and docetaxel (DTX): the latter is one of the most aggressive standard combination regimens in patients with advanced NSCLC. Patients with previously untreated stage IIIB or IV NSCLC were randomized to receive either oral S-1 80 mg/m2 twice daily on days 1–21 and CDDP 60 mg/m2 on day 8 every 5 weeks or DTX 60 mg/m2 on day 1 and CDDP 80 mg/m2 on day 1 every 3 weeks, both up to six cycles. From April 2007 to December 2008, 608 patients from 66 sites in Japan were randomized to S-1 and CDDP (n = 303) or to CDDP and DTX (n = 305). S-1 and CDDP was noninferior to CDDP and DTX in terms of overall survival (OS) (MST 16.1 versus 17.1 months, respectively; [HR] 1.013; 96.4% CI 0.837–1.227). The noninferiority design employed an upper CI limit of HR < 1.322 in this trial. A statistically significantly lower rate of febrile neutropenia (7.4% versus 1.0%), grade 3/4 neutropenia (73.4% versus 22.9%), grade 3/4 infection (14.5% versus 5.3%) and grade 1/2 alopecia (59.3% versus 12.3%) were observed in the S-1 and CDDP arm when compared with the CDDP and DTX arm. Thus, according to Japanese data, S-1 combined with CDDP is a standard first-line chemotherapy regimen for advanced NSCLC [Katakami et al. 2012].
In North America, a phase II trial of S-1 and CDDP was also conducted. The dose of S-1 was reduced to 25 mg/m2 twice daily, days 1-14, combined with 75 mg/m2 CDDP, day 1, every 3 weeks, due to difference in ethnicity as described earlier [Sandler et al. 2011]. The S-1 and CDDP regimen appeared to have a similar level of antitumor activity and toxicity when compared with that of established CDDP-based doublets for advanced NSCLC.
At the same time, S-1 combined with CBDCA was investigated in patients with advanced NSCLC in Japan. CBDCA is an analog of CDDP and produces less nonhematologic toxicity than CDDP. Many physicians prefer to use CBDCA-containing chemotherapy for the treatment of advanced NSCLC. A phase I/II study of S-1 and CBDCA combination therapy demonstrated that administration of S-1 (80 mg/m2/day) on days 1–14 in combination with CBDCA (area under the curve [AUC] of 5) on day 1 of 3-week cycles yielded similar efficacy to those of other platinum doublets. S-1 combined with CBDCA had a more favorable toxicity profile than that typically seen with platinum-based regimens, especially with regard to neutropenia, febrile neutropenia, neuropathy and alopecia [Tamura et al. 2009].
The Lung Cancer Evaluation of S-1 (LETS) study was a multicenter, randomized, phase III trial conducted by the West Japan Thoracic Oncology Group (WJTOG). This noninferiority trial of S-1/CBDCA in comparison with CBDCA/paclitaxel (PTX) combination therapy included chemotherapy-naïve patients with advanced NSCLC. From August 2006 to May 2008, 564 patients from 30 institutions were enrolled in this study. At the planned interim analysis, with a total of 268 death events available, the study passed the O’Brien–Fleming boundary of 0.0080 for a positive result and noninferiority of S-1/CBDCA compared with CBDCA/PTX was confirmed for OS (HR 0.928; 99.2% CI 0.671–1.283). MST was 15.2 months in the S-1/CBDCA arm and 13.3 months in the CBDCA/PTX arm, with 1-year survival rates of 57.3% and 55.5%, respectively (Figure 1). Rates of leukopenia or neutropenia of grade 3 or 4 and of febrile neutropenia were significantly lower (p < 0.001) whereas thrombocytopenia of grade 3 or 4 was more common (p < 0.001) for S-1/CBDCA than for CBDCA/PTX. Alopecia, neuropathy and arthralgia occurred less frequently with S-1/CBDCA (p < 0.001) [Okamoto et al. 2010].
CATS and LETS demonstrated that S-1 combined with CDDP or CBDCA was one of the standard platinum doublet regimens in the first-line setting for patients with advanced NSCLC in Japan. In the real clinical situation, many treatment options could be selected for patients with advanced NSCLC, especially non-SCC. There were several potential clinical benefits in selecting S-1 containing regimens, such as its oral administration, relative well-tolerated toxicity (especially in Japanese patients) and histology preference described below. S-1 containing regimen is selected frequently in Japan, although these potential benefits are indifferent in the treatment of NSCLC.
Recently, updated and subgroup analyses were performed to assess the efficacy and safety according to histological type (SCC or non-SCC) in the first phase III study of LETS. As first-line treatment for patients with SCC, S-1/CBDCA yielded a trend toward improved OS, with a 3.4-month improvement in median OS when compared with CBDCA/PTX (14.0 versus 10.6 months; HR 0.713; 95% CI 0.476–1.068). For patients with SCC, S-1/CBDCA produced significantly lower rates of febrile neutropenia when compared with CBDCA/PTX (4% in the S-1/CBDCA and 19% in the CBDCA/PTX group, p = 0.017) as well as lower rates of neuropathy [Yoshioka et al. 2012b]. In this regard, given the historical context of NSCLC studies focused on SCC, a survival advantage observed with S-1/CBDCA in SCC patients is promising and needs to be substantiated by further phase III studies. Given its efficacy and favorable toxicity profile, the combination of S-1 and CBDCA is also a feasible platinum-based option to which molecularly targeted agents can be added.
It is unclear whether the possible survival benefit conferred by S-1 and CBDCA in SCC patients is due to an intrinsic superiority of this drug combination compared with CBDCA and PTX, to a reduced toxicity or to other factors. S-1 and CBDCA was associated with a significantly lower rate of febrile neutropenia compared with CBDCA and PTX as well as with a lower rate of neuropathy. SCC patients in the S-1/CBDCA arm received DTX more frequently as a second-line treatment than did those in the CBDCA/PTX arm (58.2% versus 30.5%, respectively, p = 0.003), possibly because the former patients were in better condition as a result of a better-tolerated first-line regimen. The reduced toxicity of S-1/CBDCA, especially with regard to neuropathy and neutropenia, may thus have allowed for more frequent application of second-line treatment with DTX. Given the increasing number of active drugs available for second-line treatment, subsequent therapies instituted after disease progression can have a substantial impact on OS in advanced NSCLC [Hayashi et al. 2012].
Second-line treatment for advanced NSCLC
Although first-line chemotherapy is associated with improved survival and quality of life in patients with advanced NSCLC, virtually all patients eventually develop progressive disease requiring additional treatment. There are currently several agents, such as DTX, PEM and erlotinib, recognized for use in patients with metastatic NSCLC progressing after platinum-based doublet chemotherapy. The outcomes for second-line therapy remain suboptimal, with overall response rate (ORR) in this setting of less than 10%, median PFS less than 3 months, MST between 5.7 and 8.3 months, and 1-year OS between 30% and 37% [Fossella et al. 2000; Hanna et al. 2004; Shepherd et al. 2000, 2005].
S-1 monotherapy as second-line treatment was evaluated in patients with advanced NSCLC. Two prospective phase II studies of S-1 monotherapy (80 mg/m2 for 28 days, followed by 14 days of rest, every 6 weeks) were conducted in Japan. In the first study, the efficacy and toxicity of S-1 was evaluated in 50 patients with advanced NSCLC who had previously received one platinum-based chemotherapy regimen. The response rate was 12.5% (95% CI 3.1–21.9%), and median PFS and median OS were 2.5 and 8.2 months, respectively. No grade 4 toxicities were encountered and grade 3 nonhematological toxicities were observed in 10.4% [Totani et al. 2009]. In the second study, the response rate was 14% (95% CI 3.5–23.8%) for 44 evaluable patients who were treated with S-1after platinum-based chemotherapy. Median PFS and OS were 4.2 and 16.4 months, respectively [Shiroyama et al. 2011]. These data indicate that the efficacy of S-1 monotherapy as second-line chemotherapy is promising, with mild toxicity. Currently, a randomized phase III trial of S-1 when compared with DTX in patients with advanced NSCLC for second-line setting is ongoing in Eastern Asia, including in Japan, Taiwan and China (EAST-LC trial).
Combination of S-1 with a nonplatinum agent
Considering the toxicities of CDDP-based chemotherapy and the poor prognosis associated with advanced NSCLC, explorations of active and less-toxic substitutable combinations that include new, active compounds with novel mechanisms of action are needed. S-1 was also investigated when used in combination with several nonplatinum agents the in first-line or second-line setting for patients with advanced NSCLC.
S-1 combined with gemcitabine (GEM) has been studied for the treatment of advanced NSCLC or pancreatic cancer. Studies have documented a significant increase in hENT1 (a major modulator of cellular uptake of GEM) and GEM cellular uptake after S-1 or 5-FU treatment in pancreatic cancer cell lines. Significant tumor growth inhibition has been reported in mice treated with S-1 followed by GEM when compared with both untreated and S-1 and GEM concomitantly treated mice in other schedules [Nakahira et al. 2008]. This combination may result in a synergistic effect in a sequence-dependent manner. This synergistic effect, however, may be associated with increased toxicity. A phase II trial using combination therapy with S-1 and GEM in advanced pancreatic cancer demonstrated mild toxicity and favorable efficacy at the recommended doses of S-1 (60 mg/m2 on days 1–14) and GEM (1000 mg/m2 on days 8 and 15) in Japan [Nakamura et al. 2006].
A randomized phase II study of two different schedules of S-1 and GEM was evaluated in patients with NSCLC. Patients were given oral S-1 (60 mg/m2 twice daily) on days 1–14 with GEM (1000 mg/m2) on days 1 and 8 (arm A) or on days 8 and 15 (arm B). A total of 80 patients were enrolled in this trial. The response rates of arm A and arm B were 22.0% and 28.9%, respectively (p = 0.606). Median time to treatment failure was 3.6 months in arm A and 4.8 months in arm B. Median time to progression was 4.1 months in arm A and 5.5 months in arm B. The MST was 15.5 months and 18.8 months in arm A and arm B, respectively. The toxicity profile was relatively mild and did not differ very much between the two arms. The arm B regimen was selected for further studies in patients with NSCLC because it was associated with a higher response rate and survival [Satouchi et al. 2010].
Another phase II study of S-1 and GEM as second-line chemotherapy (S-1 at 60 mg/m2 twice daily, on days 1–14 and GEM at 1000 mg/m2 on days 8 and 15 repeated every 3 weeks) was conducted. Treatment was administered for a median of 4 courses (range 1–13) in 34 patients. The ORR was 23.5% (95% CI 9.1–38.0%), and the median PFS and OS were 6.6 and 19.9 months, respectively [Takiguchi et al. 2011].
In addition, S-1 combined with DTX, which are the most popular agents for second-line treatment of NSCLC, was investigated in Japan. S-1 and DTX have shown synergy in human gastric and breast cancer xenograft models [Suto et al. 2006; Wada et al. 2006]. A phase I/II study of S-1 and DTX in patients with previously treated NSCLC was conducted. Patients were treated with DTX (starting dose 40 mg/m2) intravenously on day 1 and oral administration of S-1 at a fixed dose of 80 mg/m2 on days 1–14 every 3 weeks. The recommended dose of DTX was 40 mg/m2 in combination with S-1 80 mg/m2/day. An ORR of 24.1% (95% CI 10.3–43.5%) was observed in 29 evaluable patients, and median OS and 1-year survival rate were 11.8 months and 42%, respectively. Major grade 3–4 toxicities were neutropenia (34.5%), leukopenia (20.6%) and anemia (10.3%). This data indicate that combination chemotherapy is highly active and well tolerated in patients with previously treated NSCLC [Atagi et al. 2008]. Although doublet chemotherapy as second-line treatment of advanced NSCLC is more toxic and does not improve OS when compared with single-agent therapy [Di Maio et al. 2009], randomized phase III studies investigating survival outcomes in response to S-1 combined with either GEM or DTX are warranted.
Chemotherapy-naïve patients with advanced NSCLC were treated with irinotecan (CPT-11; 150 mg/m2) on day 1 and with oral S-1 (80 mg/m2) on days 1–14 every 3 weeks. An ORR of 21.4% was observed in 56 evaluable patients, and median OS and PFS were 15.0 and 4.9 months, respectively. Hematologic toxicities of grade 3 or 4 toxicities were neutropenia (25%), thrombocytopenia (3.6%), and anemia (3.6%). The combination of S-1 and CPT-11 is a potential alternative option with favorable toxicity profile for the treatment of advanced NSCLC [Okamoto et al. 2008].
Chemoradiotherapy containing S-1 for locally advanced NSCLC
A list of studies investigating chemoradiotherapy containing S-1 and CDDP (cisplatin) for locally advanced NSCLC is given in Table 2.
Chemoradiotherapy containing S-1 and CDDP (cisplatin) for locally advanced non-small cell lung cancer (NSCLC).

bid, twice daily; DTX, docetaxel; MST, median survival time; TRT, thoracic radiotherapy.
Recommended dose for phase II study.
The standard treatment modality in patients with unresectable stage III NSCLC is concurrent chemoradiotherapy (CRT) [Pfister et al. 2004]. However, this combined treatment is associated with greater acute toxicity, including bone marrow suppression, pneumonitis and esophagitis, when compared with the sequential combination of chemotherapy and thoracic radiotherapy (TRT) [Auperin et al. 2010]. When third-generation chemotherapeutic agents, such as irinotecan, PTX, GEM and DTX were combined with concurrent TRT, the dose of the chemotherapeutic agent could be reduced greatly or the administration schedule could be modified to reduce the probability of severe adverse events [Choy et al. 1998; Takeda et al. 1999]. Therefore, induction chemotherapy with sufficient systemic doses of the agents was considered a potentially effective addition to the concurrent CRT [Vokes et al. 2002]. Nevertheless, a recent randomized trial (CALGB39801) showed that two cycles of induction chemotherapy with full doses of two cycles of CBDCA and PTX did not provide a survival benefit over concurrent CRT alone that used weekly CBDCA and PTX at lower doses [Vokes et al. 2007].
Two phase II trials of S-1 and CDDP combined with concurrent TRT were evaluated in locally advanced NSCLC in Japan. In the first trial conducted by the WJTOG, there was a high response rate of 82% and a median PFS of 20 months; median OS was not reached at a follow-up time range from 24 to 37 months [Ichinose et al. 2011]. Another trial of similar schedule and dose that was conducted during almost the same period showed an ORR of 88%, median PFS of 12 months, and a MST of 33.1 months, whereas the median follow-up time was 25 months, ranging from 12 to 38 months [Ohyanagi et al. 2009]. Chemotherapy combined with TRT using systemic doses of S-1 and CDDP has the advantage of eradicating occult distant metastases. In addition, both 5-FU and CDDP have a radiosensitizing effect in various cancers, including NSCLC, according to preclinical and clinical studies [Byfield et al. 1982; Douple and Richmond, 1982]. S-1 was given orally for 14 consecutive days in each cycle of chemotherapy, providing long-term potential radiosensitization. Furthermore, CDHP in S-1 also has a radiosensitizing effect and strong DPD activity [Fukushima et al. 2010; Takagi et al. 2010]. Although the present CRT should be acceptably safe in terms of the frequency of grade 3 and 4 adverse events, treatment-related death of two patients was observed in a WJTOG study. Therefore, clinicians should be aware that there is not completely safe regimen for use within concurrent CRT.
Two phase I studies of split-dose S-1 and CDDP combined with concurrent TRT have been published [Chikamori et al. 2009; Kaira et al. 2009]. However, this split-dose regimen using S-1 and CDDP has not been investigated for advanced NSCLC and, thus, the potential efficacy of this chemotherapy has not yet been characterized.
At present, the WJOG (which has changed its name from WJTOG since 2007) is conducting a randomized phase II trial comparing S-1 and CDDP with concurrent TRT to combination chemotherapy using CDDP and vinorelbine with concurrent TRT. Furthermore, a phase III trial of the most promising chemotherapy from that phase II trial combined with concurrent TRT versus the WJOG standard treatment of weekly CBDCA and PTX with concurrent TRT for locally advanced NSCLC is likely to be conducted in the near future [Yamamoto et al. 2010a].
In patients with marginally resectable stage III NSCLC, a small phase II trial has also reported that the safety and efficacy of induction treatment using S-1 plus CDDP and concurrent TRT and surgical resection was a feasible and promising new treatment modality [Maruyama et al. 2011].
S-1 as adjuvant chemotherapy for early stage NSCLC
Recent meta-analyses of data from many clinical studies showed that postoperative adjuvant chemotherapy improves survival in patients with early stage NSCLC [Pignon et al. 2008]. S-1 is thought to be a potentially suitable as adjuvant chemotherapy when combined with CDDP or prolonged daily use of S-1 instead of tegafur-uracil (UFT) in Japan.
WJOG recently presented the results of a randomized phase II trial of adjuvant chemotherapy with S-1 and CDDP versus S-1 alone in patients with resected stage II–IIIA NSCLC. Both S-1 monotherapy and a combination of S-1 and CDDP were feasible as adjuvant chemotherapy for completely resected NSCLC [Yoshioka et al. 2012a]. An ongoing randomized phase III trial of S-1 compared with UFT in patients with completely resected stage I NSCLC is being conducted by the Japan Clinical Oncology Group (JCOG). In the former study, the predictive molecular markers of S-1 are also identified cyclopaedically by microarray-based gene expression analysis. If molecular targets for S-1 were identified in many analyses like this, the individual therapy of S-1 could be pragmatized in the near future.
Conclusions
S-1 is an orally administered, active cytotoxic agent against NSCLC. In Japanese clinical practice, most physicians prefer to use S-1 for the treatment of NSCLC in various situations because of its oral administration, relatively well-tolerated toxicity (especially in Japanese patients) and expected comparative efficacy to other antitumor agents. S-1 combined with CDDP or CBDCA is a standard treatment regimen for use in the first-line setting. S-1 monotherapy may be used as second-line or later therapy. In patients with locally advanced NSCLC, S-1 and CDDP with concurrent TRT is associated with encouraging outcomes. Adjuvant chemotherapy with S-1 is also promising against early stage NSCLC.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The author has no conflicts of interest to declare.