
Abstract
The c-MET (mesenchymal–epithelial transition factor) pathway is dysregulated in many human cancers and promotes tumor growth, invasion and dissemination. The c-MET receptor tyrosine kinase can be activated via gene mutation, gene amplification, protein overexpression and/or a ligand-dependent autocrine/paracrine loop. Abnormalities in c-MET signaling have been reported to correlate with poor clinical outcomes and drug resistance in patients with cancer. Significant progress has been made in advancement of c-MET pathway inhibitors through to clinical trials. A robust pipeline of high-quality inhibitors targeting different aspects of c-MET activation is currently being explored in phase I, II and III clinical trials across multiple tumor types. Preliminary data demonstrate promising clinical activity with these agents, along with an acceptable toxicity profile. In this manuscript, the pharmacological profile of drugs targeting the c-MET pathway and available data from ongoing clinical trials of these drugs are discussed.
Introduction
Inhibiting c-MET (mesenchymal–epithelial transition factor) signaling is emerging as a promising strategy for a new class of targeted cancer therapies. Several c-MET inhibitors are in various stages of clinical development and have demonstrated activity in different tumor types. c-MET is a receptor tyrosine kinase encoded by the proto-oncogene MET and has a high affinity for hepatocyte growth factor (HGF; also known as scatter factor, SF) [Cooper et al. 1984]. Activation of c-MET, mediated by HGF binding, promotes several processes involved in oncogenesis, including tumor cell proliferation, migration, invasion, angiogenesis, protection from apoptosis and metastasis, working through several other signaling pathways such as PI3K/Akt, Src, STAT3, and Ras/Mek [Comoglio et al. 2008; Zhang et al. 2003; Trusolino and Comoglio, 2002; Furge et al. 2000].
The c-MET pathway is frequently dysregulated in human cancers, and aberrant c-MET signaling has been reported in a wide variety of human malignancies, including gastric, lung, colon, breast, bladder, head and neck, ovarian, prostate, thyroid and pancreatic as well as hematologic malignancies and central nervous system tumors [Liu et al. 2008; Birchmeier et al. 2003; Di Renzo et al. 2000; Ferracini et al. 1995]. Oncogenic activation of c-MET signaling can be induced by specific genetic lesions, transcriptional upregulation, ligand-dependent autocrine or paracrine mechanisms [Bean et al. 2007; Schmidt et al. 1997; Houldsworth et al. 1990]. Inherited and somatic mutations in MET have been found in papillary renal carcinoma tumor samples, providing strong direct evidence of the pathway's oncogenic potential [Jeffers et al. 1997; Schmidt et al. 1997]. In addition, there is accumulating evidence that acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors and angiogenesis inhibitors can be due, in part, to increased activation of the c-MET pathway [Ebos et al. 2009; Engelman et al. 2007]. For example, amplification of MET leads to gefitinib resistance in lung cancer by mediating HER3-dependent activation of PI3 kinase and these tumors are sensitive to c-MET inhibitors [Bean et al. 2007; Engelman et al. 2007].
Approaches to inhibiting the c-MET axis in the clinic
Several strategies have been developed to inhibit the c-MET signaling pathway in cancer, each focusing on one of the serial steps that regulate MET activation (Figure 1). These strategies include selective c-MET kinase inhibitors such as tivantinib (ARQ 197), JNJ-38877605 and PF04217903 which have specific selectivity for c-MET receptor tyrosine kinases; nonselective c-MET kinase inhibitors such as PF02341066, cabozantinib (XL184), GSK1363089, MK-2461, MP470 and MGCD265 which have broad activity against c-MET and other receptor tyrosine kinases; anti-c-MET monoclonal antibodies (MetMAb) are also selective, but bind to the receptor, leading to internalization and degradation as opposed to inhibiting tyrosine kinase activity; anti-HGF monoclonal antibodies (AMG102, SCH900105) bind to the circulating ligand, HGF; and c-MET/HGF competitors (NK4).
Schematic representation of the c-MET (mesenchymal–epithelial transition factor) dependent signaling pathway. Activation of c-MET results in the recruitment of several SH2-domain-containing signal transducers that in turn activate a number of pathways, including the GRB2–SOS–RAS–RAF–MEK–ERK axis, leading to cell proliferation and invasion [Furge et al. 2000]. Strategies to inhibit c-MET signaling have therefore targeted different steps involved in c-MET activation. Ab, antibody; HGF, hepatocyte growth factor.
In this review, an overview of c-MET pathway inhibitors will be provided, supported by available phase II clinical trial data.
Tivantinib
Pharmacological profile
Tivantinib (ARQ 197) is an oral, highly selective, non-adenosine triphosphate (ATP)-competitive c-MET inhibitor, which is now in phase III development. In a panel of 230 human protein kinases, tivantinib only selectively inhibited c-MET to an appreciable extent; this high degree of selectivity is related to its ability to decrease Vmax (The maximum activity) without affecting the Km (Michaelis constant) of ATP and suggests a non-ATP-competitive mechanism of inhibition [Munshi et al. 2010; Jeay et al. 2007]. Tivantinib activity has been assessed against c-MET in different cancer cell lines and xenograft tumor models [Gu et al. 2009], and inhibits c-MET phosphorylation and downstream signaling in different human cancer cell lines with a 50% inhibitory concentration (IC50) of 100–300 nM [Munshi et al. 2010]. The antiproliferative effect of tivantinib is related to c-MET signaling, as in c-MET null human cancer cell lines, little, if any antiproliferative effect was observed [Munshi et al. 2010]. Tivantinib inhibits c-MET receptor kinase within 24 h of administration and can be sustained for up to 8–12 h following withdrawal of tivantinib [Gu et al. 2009].
Treatment of different tumor xenograft-bearing mice with tivantinib has demonstrated significant tumor growth reductions of 45–79% in colon, gastric, breast, prostate and pancreatic cancer models [Munshi et al. 2010; Anderson et al. 2007; Li et al. 2007]. In human colon xenograft tumors, a significant reduction in c-MET autophosphorylation was observed within 24 h following single oral dose administration of tivantinib, and plasma levels of tivantinib were more than threefold above the tivantinib Ki (inhibitory constant) for c-MET at 10 h [Munshi et al. 2010]. Consistent with the role of c-MET signaling in metastasis, tivantinib has also demonstrated the ability to prevent bone metastases in mouse models of metastatic breast cancer and colon cancer [Anderson et al. 2007; Li et al. 2007].
Clinical development
Among c-MET inhibitors, tivantinib is the most advanced in clinical development. Several phase I and phase II studies have been completed and phase III trials are in process [Adjei et al. 2011a].
Phase I dose-escalation study of tivantinib in advanced solid tumors
Data from an open-label, single-center, phase I study of tivantinib in advanced solid tumors were recently reported [Yap et al. 2011]. Tivantinib was administered orally at 100–400 mg twice daily continuously in 28-day cycles. Fifty-one patients with advanced solid tumors were enrolled into sequential dose-escalation cohorts. The most common toxicities were grade 1–2 fatigue (n = 8; 15.7%), nausea (n = 7; 13.7%) and vomiting (n = 6; 11.8%). In the 400 mg twice daily cohort, a dose-limiting toxicity (DLT) of grade 3 febrile neutropenia was observed in two patients. In one of these patients, two other grade 3 DLTs (mucosal inflammation and palmar-plantar erythrodysesthesia) were also observed. All DLTs resolved within 2 weeks of tivantinib discontinuation. Data from this study recommended the use of tivantinib 360 mg twice daily in phase II studies. Mean time to maximum plasma concentration and half life for tivantinib were 2 and 5 h, respectively, and systemic exposure to tivantinib [maximum plasma concentration and area under the curve (AUC)] increased with increasing dose. Steady-state cumulative mean trough plasma concentration achieved for all dose levels of tivantinib was at 661 ng/ml (day 29 of treatment), which was well above the IC50 for in vitro c-MET inhibition of 0.3 µmol/liter (110 ng/ml). Tivantinib decreased intratumoral phosphorylated c-MET, total c-MET, phosphorylated focal adhesion kinase and increased apoptosis as shown by TUNEL assays. More than three circulating tumor cells at baseline were detected in 15 patients, eight (53.3%) of whom had more than a 30% decline in circulating tumor cells after treatment. A decline of up to 100% in circulating endothelial cell counts after treatment was observed in 25 (58.1%) patients [Yap et al. 2011; Adjei et al. 2011a]. No significant change in dynamic contrast-enhanced magnetic resonance imaging parameters were observed after 7 days of tivantinib treatment. The best treatment response in this phase I trial was stable disease (SD) for over 4 months in 14 patients (27%), with minor regressions in gastric and Merkel cell carcinomas. One patient with metastatic melanoma with T276A MET mutation experienced SD for 20 weeks and had a marked improvement in symptoms.
Phase I dose-escalation study of tivantinib in combination with sorafenib in advanced solid tumors
This study was undertaken based on the preclinical synergy of tivantinib in combination with sorafenib. The primary objective of the study was to define the maximum tolerated dose and recommended phase II dose of tivantinib in combination with sorafenib. The preliminary results were presented at the 2011 Annual Meeting of the American Society of Clinical Oncology [Adjei et al. 2011b]. Twenty-two patients were enrolled and treated at two dose levels. No DLTs were observed at the first dose level of tivantinib 360 mg twice daily plus sorafenib 200 mg twice daily. For the next cohort, dosing was increased to the full single-agent dose of both drugs: tivantinib 360 mg twice daily plus sorafenib 400 mg twice daily. One of nine patients (11.1%) at dose level 2 experienced two DLTs (grade 3 fatigue and grade 3 dyspnea), making this dose level the recommended phase II dose. The most commonly reported drug-related adverse effects of any grade were fatigue (36.4%), diarrhea (27.3%), anorexia (22.7%) and rash (22.7%). Pharmacokinetic analysis indicated that sorafenib had no effect on the disposition of tivantinib.
Among 14 of 18 patients with evaluable responses, a best response of SD for 7–32 weeks (median 12 weeks) was demonstrated [Adjei et al. 2011b]. The majority of patients with SD had renal cell cancer or hepatocellular cancer. These results indicate that a combination of sorafenib and tivantinib is safe and may have therapeutic potential.
Phase I dose-escalation study of tivantinib in combination with gemcitabine in advanced solid tumors
This ongoing multicenter, phase Ib dose-escalation trial is examining the safety and tolerability of tivantinib at doses of 120–360 mg twice daily across different schedules (continuous versus continuous with 1 week break every 2 or 3 weeks) in combination with gemcitabine at 1000 mg/m2/weekly × 3 every 4 weeks [Camacho et al. 2011].
As of January 2011, a total of 32 patients with metastatic breast (12), ovarian (14), and uterine (6) carcinoma were enrolled and treated. No DLTs were observed. The most commonly observed adverse effects were thrombocytopenia (66%), anemia (66%), neutropenia (63%), fatigue (34%), nausea (31%), and leukopenia (13%). Treatment-related serious adverse effects were observed in three patients (pancytopenia, thrombocytopenia, hypotension and interstitial lung disease, grade 3) [Camacho et al. 2011]. Among the 27 patients with evaluable responses, five (two breast, one uterine, two ovarian) had partial response (PR, response rate 19%), and 15 had decline (4–87%) in tumor markers (CA15.3, CA125, CEA). Two patients (with ovarian and breast carcinoma) with PR and two with SD (with breast carcinoma) had failed to respond to prior gemcitabine. On the basis of the favorable safety profile and encouraging signs of antitumor activity, phase II combination studies are being planned in different tumor types.
Randomized, placebo-controlled phase I/II study of tivantinib, irinotecan and cetuximab in patients with wild-type KRAS metastatic colorectal cancer who received front-line systemic therapy
This study is based on the hypothesis that adding tivantinib to irinotecan plus cetuximab may decrease resistance to cetuximab treatment and improve patient outcomes. Patients with locally advanced or metastatic colorectal cancer who received more than one prior line of chemotherapy, were KRAS wild type and had Eastern Cooperative Oncology Group performance status less than 2 were included in this study [Bessudo et al. 2011]. Patients were treated with irinotecan (180 mg/m2) and cetuximab (500 mg/m2) every 2 weeks along with escalating doses of tivantinib (120, 240, 360 mg) twice daily. Preliminary toxicity and efficacy data are available for nine patients. No DLTs were observed and grade 3/4 adverse events included neutropenia (grade 4 in one patient), fatigue (grade 3 in two patients) and one case each of grade 3 leukopenia, acneiform rash, vomiting, diarrhea, anemia and syncope. In nine patients with evaluable responses, best responses included one complete response (CR) (after four cycles), 2 PRs (after two cycles), five SD and one progressive disease [Bessudo et al. 2011]. The randomized phase II portion of the study continues to accrue data for the recommended phase II dose of 360 mg tivantinib twice daily.
Phase II combination study of tivantinib plus erlotinib versus erlotinib plus placebo in metastatic non-small cell lung cancer
A multicenter, randomized, placebo-controlled, double-blind phase II study designed to compare treatment with tivantinib plus erlotinib with erlotinib plus placebo in patients with inoperable, locally advanced/metastatic non-small cell lung cancer (NSCLC) was recently completed (Figure 2) [Schiller et al. 2010]. This study enrolled patients who had received one prior chemotherapy regimen (other than an EGFR inhibitor) for NSCLC. Eligibility criteria included confirmed availability of archival tissue suitable for analysis of KRAS, EGFR, and c-MET. Eligible patients (n = 167) were randomly assigned to receive either erlotinib 150 mg once daily plus tivantinib 360 mg twice daily (n = 84) or erlotinib 150 mg once daily plus placebo twice daily (n = 83) in a 28-day cycle [Schiller et al. 2010].
Study design of the phase II study of tivantinib and erlotinib in patients with advanced non-small cell lung cancer (NSCLC) [Schiller et al. 2010]. BID, twice daily; EGFR, epidermal growth factor receptor; ORR, overall response rate; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PO, oral; QD, once daily; TKI, tyrosine kinase inhibitor; US, United States.
Results of the phase II study of tivantinib and erlotinib in patients with advanced non-small cell lung cancer.

CI, confidence interval; DCR, disease control rate; HR, hazard ratio; ITT, intention to treat; OS, overall survival; PFS, progression-free survival; PR, partial response; SD, stable disease.
Of patients with an evaluable response, PRs were observed in seven of 73 (10%) in the tivantinib plus erlotinib arm compared with five of 72 (7%) in the erlotinib plus placebo arm. Disease control rates were 66% and 54% respectively (Table 1) [Schiller et al. 2010].
Interestingly, this study also demonstrated the potential antimetastatic activity of tivantinib. For intention-to-treat patients, median time to new metastatic lesions was increased from 3.6 months in the erlotinib plus placebo arm to 7.3 months in the tivantinib plus erlotinib arm (HR 0.49; 95% CI 0.31–0.78). Patients with nonsquamous histology had an even more pronounced effect, with median time to metastatic disease being increased from 3.6 to 11.0 months (HR 0.46; 95% CI 0.26–0.82) (Table 1) [Sequist et al. 2010].Overall, treatment with tivantinib was well tolerated with no significant differences in adverse effects between treatment and control arms. The most frequent adverse effects included grade 1/2 rash, diarrhea, anorexia, anemia and fatigue [Schiller et al. 2010]. Based on the results of this study, a global phase III randomized, double-blind, placebo-controlled study of tivantinib plus erlotinib in previously treated patients with metastatic nonsquamous NSCLC is currently ongoing (Table 4).
MetMAb
Pharmacological profile
MetMAb is a monovalent monoclonal antibody directed against c-MET, which prevents HGF from binding to the c-MET receptor, thereby blocking HGF-induced dimerization and receptor activation. Attempts to inhibit c-MET signaling using monoclonal antibodies have been challenging because most antibodies have intrinsic agonistic activity and single antibodies have been unable to completely block the SF/HGF:c-MET binding [Cao et al. 2001; Ohashi et al. 2000; Prat et al. 1998]. Recently, a one-armed variant of the anti-c-MET antibody 5D5, MetMAb, was developed to avoid agonistic activity that can occur when divalent antibodies bind and crosslink MET receptors. MetMAb binds to the Sema domain of c-MET, a region which is critical for binding HGF [Nguyen et al. 2003].
MetMAb inhibited c-MET tyrosine phosphorylation, cell proliferation, migration, and apoptosis in U87 glioblastoma cells, strongly driven by autocrine or paracrine SF/HGF-c-MET signaling [Martens et al. 2006]. Treatment of the orthotopic model of U87 and G55 tumors with MetMAb significantly inhibited growth only in SF/HGF-activated tumors. In addition, in MetMAb-treated tumors, cell proliferation was reduced more than 75%, microvessel density was reduced more than 90% and apoptosis was increased more than 60%. In a c-MET- and HGF-expressing, autocrine-driven, human KP4 pancreatic cancer orthotopic model, MetMAb also significantly inhibited c-MET phosphorylation, with a concomitant decrease in tumor growth and improvement in survival [Jin et al. 2008].
Phase I study of MetMAb in combination with bevacizumab in patients with advanced solid malignancies
The combination of MetMAb with bevacizumab was tested in a phase I study which consisted of three parts: 3 + 3 dose escalation of MetMAb evaluating 1, 4, 10, 15, 20, and 30 mg/kg intravenously every 3 weeks; expansion at 15 mg/kg intravenously every 3 weeks; and combination of MetMAb at 10 and 15 mg/kg plus bevacizumab 15 mg/kg intravenously every 3 weeks. Baseline and post-treatment serum was collected for evaluation of pharmacodynamic biomarkers possibly affected by inhibition of c-MET and/or vascular endothelial growth factor (VEGF) signaling [Moss et al. 2010, 2011].
A total of 43 patients (21 in escalation, 13 in expansion, nine in combination) were treated [Moss et al. 2010, 2011]. The most frequently observed toxicities were fatigue, peripheral edema and hypoalbuminemia. No grade 3–5 treatment-related adverse events were reported with the combination; a grade 1 and DLT of hemoptysis was reported in one patient with central necrosis of pulmonary metastases. There were no pharmacokinetic interactions with bevacizumab, and MetMAb had a half life of 11 days. CR was observed in one patient with gastric carcinoma after four cycles of single-agent MetMAb. The combination of MetMAb with bevacizumab was safe and well tolerated. A phase II trial of MetMAb in combination with bevacizumab plus paclitaxel in patients with triple-negative breast cancer is currently ongoing (Table 4).
Phase II study evaluating MetMAb in combination with erlotinib in patients with advanced NSCLC
In a randomized, double-blind phase II study, MetMAb 15 mg/kg intravenously plus erlotinib was compared with erlotinib plus placebo in 128 patients with advanced NSCLC (Figure 3) [Spigel et al. 2011]. The study included patients with all histologies following at least one chemotherapy-containing regimen for stage IIIB/IV disease. Patients in the control arm had the option of being unblinded and crossing over to receive MetMAb after disease progression. Immunohistochemistry (IHC) was performed for c-MET in 121 patients. Those patients whose tumors stained 2+ or 3+ were defined as ‘MET high’, whereas those with either no expression or 1+ expression were defined as ‘MET low’. Archival tissue was evaluable for EGFR and KRAS mutations in 112 patients. Both treatment groups were well balanced with respect to molecular genotype and 54% of patients were c-MET-positive, which was associated with a poorer outcome (overall survival HR 2.52, placebo plus erlotinib cohort).
Study design of the phase II study of MetMAb and erlotinib in patients with advanced non-small cell lung cancer (NSCLC) [Spigel et al. 2011]. ITT, intention-to-treat; IV, intravenous; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PS, performance status; QD, once daily; q3w, every 3 weeks.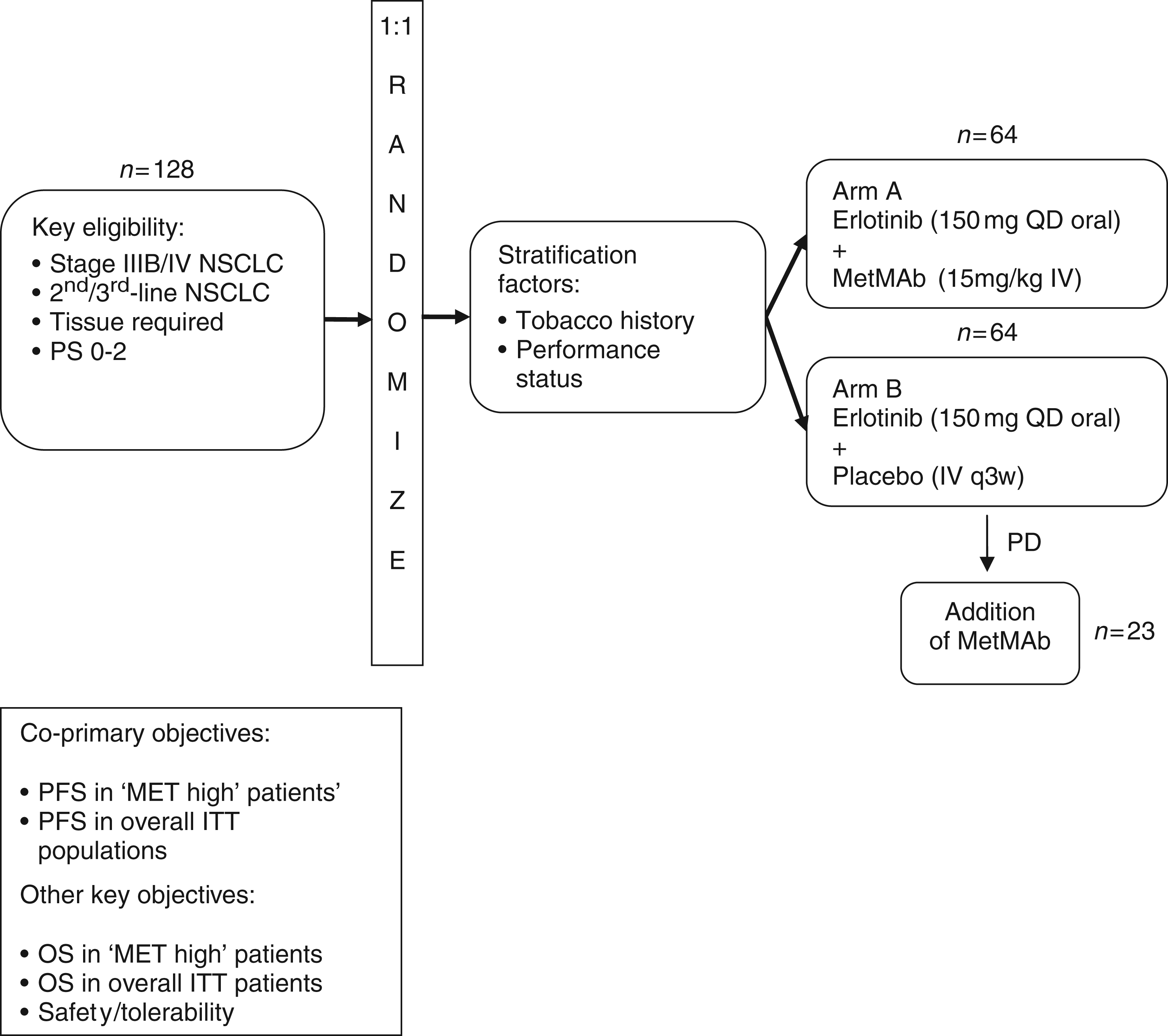
Results of the phase II study of MetMAb and erlotinib in patients with advanced non-small cell lung cancer [Spigel et al. 2011].

Initial data cut.
CI, confidence interval; C-MET, mesenchymal–epithelial transition factor; EGFR, epidermal growth factor receptor; FISH, fluorescent in situ hybridization; HR, hazard ratio; IHC, immunohistochemistry; ITT, intention to treat; OS, overall survival; PFS, progression-free survival; wt, wild type.
Foretinib
Pharmacologic profile
Foretinib (formerly XL880) is an oral multi-kinase inhibitor developed to target c-MET and several other receptor tyrosine kinases involved in tumor angiogenesis. It has a nanomolar IC50 for in vitro and in vivo inhibition of c-MET and VEGF receptor-2 (VEGFR-2), together with high in vitro affinity for platelet-derived growth factor receptor-β, Tie-2, RON, Kit, and FLT3 kinases [Qian et al. 2009]. Foretinib is an ATP-competitive inhibitor and binds deeply in the ATP pocket of both c-MET and VEGFR-2 tyrosine kinase domains with high affinity. In xenograft models of human cancers, treatment with foretinib caused necrosis and hemorrhage within 2–4 h of treatment and maximum tumor response (CR) was achieved at 96 h following five daily doses. Peak plasma concentrations after a single daily oral dose were 1–3 µmol/liter [Qian et al. 2009].
Phase I study of foretinib in patients with advanced solid tumors
In a phase I, nonrandomized, dose-finding study, patients with metastatic or unresectable solid tumors refractory to standard chemotherapy received foretinib for 5 consecutive days, every 14 days [Eder et al. 2010]. Most frequently reported treatment-related adverse events were grade 1/2 hypertension, proteinuria and fatigue. Elevation in aspartate transaminase (AST) occurred in 10 patients (25%), with one grade 3 event. Three patients had study drug discontinuation due to treatment-related adverse events, which included grade 3 elevated lipase, grade 3 tumor hemorrhage and grade 4 hemorrhage into central nervous system metastasis. At the maximum tolerated dose (3.6 mg/kg), mean Cmax and AUC0-24 values were 90.5 ng/ml (0.14 µmol/l) and 1300 ηg·h/ml (2.05 µmol/l·h) on day 1. On day 8, mean Cmax and AUC0-24 increased to 218 ηg/ml (0.34 µmol/l) and 4050 ηg·h/ml (6.40 µmol/l·h after the administration of five consecutive daily doses). The median half life across all cohorts was approximately 40 h and Tmax was approximately 4 h on both days 1 and 8.
Three patients with melanoma, medullary thyroid cancer and triple-negative breast cancer had tumor biopsies for pharmacodynamic assessment of target inhibition and downstream pathway modulation. Total c-MET and total RON were unchanged; however phosphorylated c-MET and RON were reduced in the tumors of all three patients. A decrease in downstream signaling of pERK and pAkt was also observed, together with a marked decrease in proliferation and am increase in apoptosis, measured by Ki67 and TUNEL staining of tumor cells [Eder et al. 2010].
Confirmed PRs were seen in two patients with papillary renal carcinoma and one patient with medullary thyroid carcinoma. Both patients with papillary renal carcinoma who had received no prior systemic therapy had a PR of more than 48 and 12 months, respectively. SD was observed in 22 patients (55%) [Eder et al. 2010].
Cabozantinib
Pharmacologic profile
Cabozantinib (XL184) is an oral, potent tyrosine kinase inhibitor that blocks c-MET, VEGFR2, AXL. KIT, TIE2, FLT3, and RET signaling. In the RIP-Tag2 transgenic mouse model of pancreatic neuroendocrine carcinoma, selective inhibition of VEGF reduced tumor growth but increased invasion, whereas treatment with cabozantinib decreased tumor growth, invasion, and metastasis leading to increased survival [Sennino et al. 2009].
Phase I study of cabozantinib in patients with advanced malignancies
Cabozantinib was administered on two different schedules of days 1–5 (5 and 9 schedule) or continuously on a daily basis [Salgia et al. 2008]. Fifty-five patients were treated at 13 different dose levels. DLTs included one report each of grade 3 palmar/plantar erythema, grade 3 AST, alanine aminotransferase and lipase elevations, as well as grade 2 and 3 mucositis. Other frequent treatment-related adverse events were diarrhea and hypopigmentation of the hair. Data suggested linear pharmacokinetics with a terminal half life of 59–136 h. Three patients with medullary thyroid cancer and one patient with neuroendocrine carcinoma had a PR, while SD was observed in 20 patients, which lasted for more than 6 months in 12 of these patients. Pharmacodynamic assessment of plasma samples showed a trend towards increased VEGF-A, placenta growth factor, and reduced soluble VEGFR-2 levels.
Phase Ib/II study of cabozantinib with and without erlotinib in patients with NSCLC
Fifty-four patients with NSCLC with previously treated advanced NSCLC received different combinations of cabozantinib and erlotinib in a 3 + 3 design (75 mg cabozantinib plus 150 mg erlotinib, 50 mg cabozantinib plus 150 mg erlotinib, 75 mg cabozantinib plus 100 mg erlotinib, 125 mg cabozantinib plus 100 mg erlotinib, and 125 mg cabozantinib plus 50 mg erlotinib) [Wakelee et al. 2010]. Twelve patients experienced at least one DLT: diarrhea, elevated AST, palmar-plantar erythrodysesthesia, mucositis, hypertension, hypokalemia, elevated lipase, and fatigue. The most frequent adverse events were grade 3/4 diarrhea (26%), fatigue (15%), dyspnea (12%), and hypoxia (9%). No drug interaction was found in the preliminary pharmacokinetic analysis. Three patients with prior erlotinib treatment had a reduction of at least 30% in tumor measurements. One of these patients had c-MET amplification. Prolonged SD for at least 4 months was observed in some patients, including one patient with EGFR T790M mutation [Wakelee et al. 2010].
Phase II randomized discontinuation trial of cabozantinib in advanced solid tumors
A phase II study evaluated the activity of cabozantinib in patients with breast, gastric/gastroesophageal junction, small cell lung, non-small cell lung, ovarian, pancreatic, hepatocellular or prostate cancers, or melanoma [Schöffski et al. 2010]. The study consisted of two stages: a lead-in stage (stage 1) and a double-blind randomized stage (stage 2) (Figure 4). For the lead-in stage, all patients received 100 mg of cabozantinib daily for 12 weeks. At the end of stage 1, patients with CR/PR continued to receive the same dose of cabozantinib, patients with progressive disease discontinued treatment and those with SD were randomized 1:1 to receive cabozantinib in stage 2 until disease progression. Patients randomized to placebo could cross over to cabozantinib upon progression. Efficacy endpoints were overall response rate at 12 weeks in stage 1 and PFS in stage 2, with early stopping rules in the lead-in stage to project futility.
Randomized discontinuation study design for the phase II trial of cabozantinib in advanced solid tumors [Schöffski et al. 2010].
Updated outcome data from a randomized discontinuation study of cabozantinib in advanced solid tumors [Schöffski et al. 2010].

Partial response (PR) includes unconfirmed PRs (uPRs) and confirmed PRs (cPRs) at any timepoint.
PR = cPR + uPR.
Disease control rate (DCR) = [PR + stable disease (SD)] at week 12/response evaluable.
GEJ, gastro-esophageal junction; HCC, hepatocellular carcinoma; NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer.
Completed and ongoing clinical trials with c-MET (mesenchymal–epithelial transition factor) inhibitors listed in Clinicaltrials.gov.


In the melanoma cohort, 24 patients had evaluable responses: one patient achieved a PR and 11 patients achieved SD. The overall disease control rate was 50% at week 12 [Nechushtan et al. 2010]. A total of 12 patients with hepatocellular cancer and a Child–Pugh score of A whose disease had failed to respond to up to one prior treatment regimen were enrolled: seven patients had evaluable responses and, of these, two patients achieved a PR and five patients achieved SD. The overall disease control rate was 88% at 12 weeks [Van Cutsem et al. 2010].
The preliminary results from a cohort of patients with castration-resistant prostate cancer were presented at the 2011 Annual Meeting of the American Society of Clinical Oncology [Hussain et al. 2011]. Accrual was halted at 168 and patients were unblinded due to high rates of observed clinical activity. Out of 100 patients with an evaluable response in the lead-in stage, 47% had visceral disease, 78% had bone metastasis, and 47% were docetaxel pretreated. The most frequent treatment-related grade 3/4 adverse events were fatigue (11%), hypertension (7%), and hand-foot syndrome (5%). Objective tumor shrinkage occurred in 84% of patients. The overall response rate at week 12 was 5%. Prostate-specific antigen changes were not related to clinical activity. The overall disease control rate (PR + SD) at 12 weeks was 71%. Patients with bone metastases (56 of 65, 86%) had either complete or partial resolution of lesions on bone scan as early as week 6. In 28 patients receiving narcotics for bone pain, 64% had improved pain and 46% decreased or discontinued narcotics. Measures of osteoclast and osteoblast activity, and plasma C-telopeptide declined at least 50% in 55% of patients and serum total alkaline phosphatase declined at least 50% in 56% of patients [Hussain et al. 2011].
In the ovarian cancer cohort, a total of 21 patients with epithelial ovarian cancer, primary peritoneal or fallopian tube cancer with measurable disease were enrolled. Out of seven patients with evaluable responses, three achieved an unconfirmed PR and four achieved SD. The most frequently observed adverse events were rash, palmar-plantar erythrodysesthesia syndrome, pruritus, pulmonary embolism and staphylococcal infection.
To date, 397 patients with different tumor types have been enrolled. Interim data for all tumor cohorts are summarized in Table 3.
Conclusions
Preclinical studies strongly suggest abnormal c-MET signaling in many cancers, with data supporting targeting of this pathway for cancer intervention. There are various inhibitors in clinical development targeting different steps of c-MET activation. Many of these agents have demonstrated clinical activity in both phase I and II clinical trials and are being evaluated in several ongoing trials in a variety of tumor types (Table 4). Most studies have demonstrated favorable safety profiles for these agents, when used alone or in combination with other targeted agents. Of particular clinical interest, the data demonstrate activity of c-MET inhibitors in EGFR-resistant tumors and an increase in time to new metastasis. Inhibitors targeting multiple pathways, such as cabozantinib (VEGF, RET, and c-MET) may have more clinical activity across a wide spectrum of tumor types. Selective inhibitors may have activity in c-MET-driven tumors. Combinations of these selective inhibitors and other agents such as EGFR tyrosine kinase inhibitors and VEGF inhibitors may be necessary for broader activity. The results of ongoing and planned clinical trials will shed more light on the tumor types that would benefit most from these agents, which biomarkers to use for prediction of clinical activity (c-MET amplification/overexpression/mutation) and which combinations of c-MET-inhibiting drugs with other agents are likely to be more effective.
Footnotes
Acknowledgments
Matthew Joynson, a medical writer, assisted with the styling of this manuscript. The authors wrote and revised the main draft of the article.
Funding
Editorial assistance was funded by Daiichi Sankyo Europe GmbH.
Conflict of interest statement
Dr Alex Adjei has received honoraria from Daiichi Sankyo Europe GmbH for speaking at scientific symposia. Dr Neelesh Sharma declares no conflict of interest.