
Abstract
The receptor tyrosine kinase c-MET and its ligand, hepatocyte growth factor (HGF), regulate multiple cellular processes that stimulate cell proliferation, invasion and angiogenesis. This review provides an overview of the evidence to support c-MET or the HGF/c-MET signaling pathway as relevant targets for personalized cancer treatment based on high frequencies of c-MET and/or HGF overexpression, activation, amplification in non-small cell lung carcinoma (NSCLC), gastric, ovarian, pancreatic, thyroid, breast, head and neck, colon and kidney carcinomas. Additionally, the current knowledge of small molecule inhibitors (tivantinib [ARQ 197]), c-MET/HGF antibodies (rilotumumab and MetMAb) and mechanisms of resistance to c-MET-targeted therapies are discussed.
Introduction
The MET proto-oncogene encodes for the receptor tyrosine kinase (RTK), c-MET. Cells of epithelial–endothelial origin widely express c-MET, where it is essential for embryonic development [Brand-Saberi et al. 1996; Heymann et al. 1996; Bladt et al. 1995] and tissue repair [Borowiak et al. 2004; Huh et al. 2004]. Hepatocyte growth factor (HGF) is the only known ligand for the c-MET receptor and is expressed mainly in cells of mesenchymal origin, although some epithelial cancer cells appear to express both HGF and c-MET [Ma et al. 2008; Tsao et al. 2001, 1998; To and Tsao, 1998; Tuck et al. 1996; Furukawa et al. 1995]. Under normal conditions c-MET dimerizes and autophosphorylates upon ligand binding, which in turn creates active docking sites for proteins that mediate downstream signaling leading to the activation of the mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)-AKT, v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (SRC), signal transducer and activator of transcription (STAT) signaling pathways [Organ et al. 2011; Trusolino et al. 2010; Seiden-Long et al. 2008; Peschard and Park, 2007]. Such activation evokes a variety of pleiotropic biological responses leading to increased cell growth, scattering and motility, invasion, protection from apoptosis, branching morphogenesis, and angiogenesis [Sierra et al. 2008; Conrotto et al. 2005; Yi and Tsao, 2000; Silvagno et al. 1995]. However, under pathological conditions improper activation of c-MET may confer proliferative, survival and invasive/metastatic abilities of cancer cells [Benvenuti and Comoglio, 2007; Danilkovitch-Miagkova and Zbar, 2002]. This review article discusses the evidence for the potential role of c-MET as a biomarker and therapeutic target in cancer, especially non-small cell lung carcinoma (NSCLC).
Role of c-MET in cancer
c-MET was first identified in the early 1980s as the product of a chromosomal rearrangement after treatment with the carcinogen N-methyl-N′-nitro-N-nitrosoguanidine [Cooper et al. 1984]. This rearrangement results in a constitutively fused oncogene, TPR-MET, which translates into an oncoprotein following dimerization by a leucine-zipper motif located in the TPR moiety. This provides the structural requirement for c-MET kinase to be constitutively active. TPR-MET has the ability to transform epithelial cells [Rodrigues and Park, 1993; Park et al. 1986], and to induce spontaneous mammary tumors when ubiquitously overexpressed in transgenic mice [Liang et al. 1996]. These findings set the starting point for a currently ongoing effort to unveil all oncogenic abilities of c-MET. It took more than a decade to provide the proof of concept for the role of c-MET in human cancers, which became evident following the identification of activating point mutations in the germline of patients affected by hereditary papillary renal carcinomas [Olivero et al. 1999; Schmidt et al. 1999]. However, spontaneously occurring oncogenic MET mutations remain rare at 2–3% [Schmidt et al. 1999, 1997].
c-MET and hepatocyte growth factor (HGF) are highly expressed in a variety of carcinomas.

MET gene mutation and amplification
As mentioned, somatic mutations on the MET gene are rarely found in patients with nonhereditary cancer. To date, missense mutations and single nucleotide polymorphisms (SNPs) have been found in the SEMA and juxtamembrane domain of MET, whereas, activating mutations have been described mainly in NSCLC, hereditary and spontaneous renal carcinomas, hepatocellular carcinomas, gliomas, gastric, squamous cell carcinoma of the head and neck, and breast carcinomas [Stella et al. 2011; Seiwert et al. 2009; Ma et al. 2008; Giordano et al. 2000; Lee et al. 2000; Park et al. 1999]. Potentially oncogenic mutations involve mainly point mutations that generate an alternative splicing encoding a shorter protein that lacks exon 14, which encodes for the juxtamembrane domain of c-MET [Ma et al. 2008; Lutterbach et al. 2007]; point mutations in the kinase domain that render the enzyme constitutively active [Giordano et al. 2000]; and Y1003 mutations that inactivate the Cbl binding site leading to constitutive c-MET expression [Kong-Beltran et al. 2006; Peschard et al. 2001; Vigna et al. 1999]. In contrast, several other point mutations (i.e. N375S, R988C and T1010I) have been reported as SNPs since they have been found to lack transforming abilities [John et al. 2011; Tyner et al. 2010; Tengs et al. 2006].
Types of cancers where c-MET/hepatocyte growth factor (HGF) has been reported to be important in primary tumor or metastatic progression.

NSCLC, non-small cell lung carcinoma.
c-MET protein overexpression
Over the years many groups have established that c-MET and HGF are highly expressed in a large number of solid and soft tumors (for a comprehensive list, see www.vai.org/met). The list of tumors in which c-MET is expressed is large, and it has been shown that high levels of c-MET can lead to the constitutive activation of the enzyme, as well as rendering cells sensitive to subthreshold amounts of HGF. Although many of these studies have not identified the level of c-MET receptor activity/phosphorylation or compared the expression level with its normal counterpart, it could be speculated that it is expressed with autocrine loops of HGF/c-MET which increase cell proliferation and metastases [Navab et al. 2009; Yi and Tsao, 2000; Yi et al. 1998; Rong et al. 1994; Tsao et al. 1993]. Furthermore, independent studies have shown that HGF is expressed ubiquitously throughout the body, showing this growth factor to be a systemically available cytokine as well as coming from the tumor stroma [Vuononvirta et al. 2009; Parr et al. 2004; Aguirre Ghiso et al. 1999]. A positive paracrine and autocrine loop of c-MET activation can therefore lead to further MET expression [Boccaccio et al. 1994].
c-MET as prognostic markers
Reports that have considered c-MET and hepatocyte growth factor (HGF) as prognostic markers.
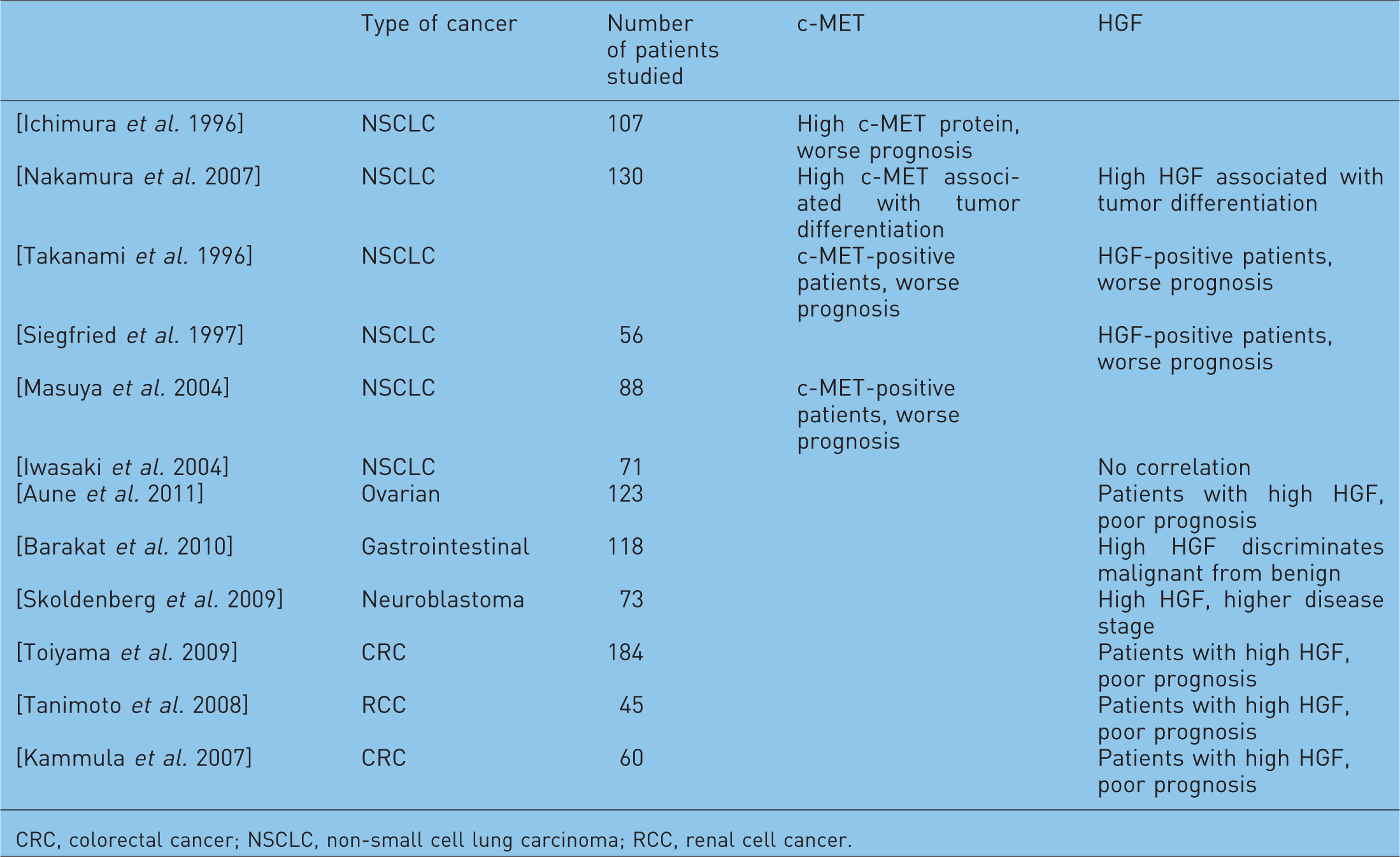
CRC, colorectal cancer; NSCLC, non-small cell lung carcinoma; RCC, renal cell cancer.
Plexins: modulators of c-MET activation
In the last decade several groups have reported that Plexin B family members (Plexin-B1, Plexin-B2, and Plexin-B3) can bind to both HGF RTK family members: c-MET and RON [Conrotto et al. 2004; Giordano et al. 2002]. B-Plexins are single-pass transmembrane receptors shown to share structural similarities with c-MET [Tamagnone and Comoglio, 2000; Tamagnone et al. 1999]. Giordano and colleagues have demonstrated that activation of Plexin-B1 by its high-affinity ligand, Sema4D, can transactivate c-MET's invasive growth program, thus promoting tumor growth, invasion, migration and angiogenesis [Sierra et al. 2008; Conrotto et al. 2005, 2004; Giordano et al. 2002]. Similarly, Swiercz and colleagues [2008, 2004] observed that the Sema4D/Plexin-B1 complex can transactivate ERBB2 promoting tumor growth [Swiercz et al. 2008, 2004].
As with other semaphorins and plexins, Plexin-B1's roles in cancer have been contradictory [Ch'ng and Kumanogoh, 2010; Capparuccia and Tamagnone, 2009]. Sema4D/Plexin-B1 complex has been associated with potentiating tumor progression in gastric [Schroeder, 1992], breast [Sierra et al. 2008; Swiercz et al. 2008], lung [Basile et al. 2006], head and neck [Basile et al. 2006, 2005], cervical [Qiang et al. 2011], ovary [Valente et al. 2009] and pancreatic [Kato et al. 2011] cancers and tumor-associated macrophage proangiogenic effects [Sierra et al. 2008]. Furthermore, Plexin-B1 point mutations found in prostate cancer bone metastases and lymph node metastases of other primary cancers render cells more motile with an increase in invasion, adhesion, and lamellipodia extension [Wong et al. 2007]. Conversely, Plexin-B1 has been shown to play a tumor suppressor role in melanomas. Two different groups have shown that Plexin-B1 expression in melanomas reduces BRAF signaling pathways [Argast et al. 2009] and decreases c-MET expression levels [Stevens et al. 2010]. These results challenge the previously published murine model lacking Plexin-B1, which demonstrated that tumor growth or angiogenesis of human melanoma cells growing in Plexin-B1-lacking environments are not affected [Fazzari et al. 2007].
c-MET as a mechanism of resistance to epidermal growth factor receptor therapies
During the past 5 years, c-MET has gained considerable interest following the report that lung adenocarcinoma cell line HCC827 (bearing the sensitizing mutation in the epidermal growth factor gene) developed resistance by amplification of the MET gene when exposed to increasing concentrations of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), erlotinib, for long periods of time [Engelman et al. 2007]. This finding was further supported by clinical evidence showing lung tumors from four EGFR TKI refractory patients displayed the MET amplification [Engelman et al. 2007]. Engelman's report was also the first to demonstrate that patients could bear two different mechanisms of resistance to an EGFR TKI, as one of the four patients harbored both the MET amplification and the commonly found gatekeeper mutation in EGFR (T790M). The latter has been shown to decrease the sensitivity of the EGFR kinase to the reversible TKI [Yun et al. 2008]. Furthermore, cells with amplified MET were now sensitive to a dual treatment with EGFR and c-MET TKI, suggesting that inhibition of both receptors could result in disease stabilization.
Reports that analyzed MET gene copy number (GCN) in non-small cell lung carcinoma (NSCLC).

c-MET activation may bypass EGFR TKIs in sensitive cells by two putative mechanisms: c-MET autophosphorylation creates docking sites where downstream signaling proteins can transduce prosurvival signals via the MAPK and PI3K/AKT signaling pathways; and transphosphorylation of other ERBB receptors may amplify the protumorigenic invasive program of c-MET. The latter mechanism is also known as ‘cross talk’ between RTKs. For all these properties, c-MET is believed to play a significant role in tumor progression and metastasis.
c-MET as a therapeutic target
Because of its pleotrophic role in cellular processes important in oncogenesis and cancer progression, c-MET is considered to be an important target in anticancer therapy [Trusolino et al. 2010; Migliore and Giordano, 2008; Peschard and Park, 2007; Corso et al. 2005]. Several molecules targeting c-MET have recently been evaluated in early phase clinical trials. Most of them are small kinase inhibitors, while some are biological antagonists and monoclonal antibodies targeting either the ligand or the receptor [Eder et al. 2009; Comoglio et al. 2008; Toschi and Janne, 2008].
Preclinical studies have shown that in animal models, the inhibition of c-MET or neutralization of its ligand impairs tumorigenic and metastatic properties of cancer cells [Corso et al. 2008; McDermott et al. 2007; Zou et al. 2007; Petrelli et al. 2006; Smolen et al. 2006; Michieli et al. 2004]. Based on this evidence, compounds that abrogate the kinase activity of c-MET have been developed [Porter, 2010]. An important issue relevant to the development of c-MET inhibitors is the identification of a molecular profile predictive of tumors that would benefit from this targeted therapy. Several studies have shown that certain gastric cancer and NSCLC cell lines display an exquisite sensitivity to c-MET inhibitors. Collectively, these studies have analyzed large panels of cell lines with known MET gene copy number and mutation variations. These studies demonstrated that cell lines with activated HGF/c-MET autocrine loop or MET amplification upon treatment with a c-MET TKI undergo apoptosis both in vitro and in vivo [Pan et al. 2010; Corso et al. 2008; Vigna et al. 2008; Lutterbach et al. 2007; Petrelli et al. 2006; Smolen et al. 2006]. The results have identified a subset of tumors based on genetic alterations that appear to be dependent on sustained c-MET activity for their growth and survival, and treatment with a single agent may inhibit tumor growth and induce cell death.
To date, only a few c-MET inhibitors have entered clinical trials. The non-ATP competitive c-MET inhibitor, tivantinib (ARQ 197), has recently completed a phase II clinical trial and shown to produce an increased response rate and overall survival when combined with erlotinib [Schiller et al. 2010]. Prior to this study, a phase I trial showed that 27% (14 out of 51 patients) of patients had stable disease for over 4 months [Yap et al. 2011]. Based on these results, tivantinib has entered a randomized, double-blind, placebo-controlled phase III study in previously treated patients with metastatic NSCLC [DeJager, 2010]. Cabozantinib (XL184), a multikinase inhibitor that targets c-MET, VEGFR2, AXL, KIT, TIE2, FLT3, and RET, has reached phase II/III trials showing reduction of tumor mass in almost 60% of patients treated with glioblastoma and an overall disease control rate of almost 50% in all of the patients who received this inhibitor in phase II studies [Wen, 2010; Salgia et al. 2008]. Cabozantinib has entered a combination regimen with erlotinib as a phase Ib/II trial. Very little is known about the results of this trial, with the exception of a partial response by patients who have MET amplification or EGFR T790M mutation [Wakelee et al. 2010]. Foretinib (XL880) is a multikinase inhibitor that targets c-MET and VEGFR2 at nanomolar concentrations [Qian et al. 2009]. It was found to stabilize the disease in 55% of the patients treated in a phase I trial [Eder et al. 2010] and, similar to other c-MET inhibitors, it recently started a phase II trial that considers the combination of this inhibitor with erlotinib [Seymour, 2010]. Lastly, the dual c-MET and ALK inhibitor, crizotinib (PF02341066), is being studied in a phase III randomized, double-blind, placebo-controlled study following several articles that demonstrated its tumor and metastasis inhibitory effects in both c-MET and ALK-positive patients [Kijima et al. 2011; Kimura et al. 2011; Ou et al. 2011]. This drug has also started a combination phase I study that considers the combination of this multikinase c-MET inhibitor with the irreversible pan-HER inhibitor PF00299804. Other compounds such as JNJ38877605 are in early phase clinical trials, but little is known regarding their results. Several reviews have been published on the subject of c-MET TKI in clinical trials [Eder et al. 2009; Comoglio et al. 2008; Toschi and Janne, 2008].
Another type of c-MET-targeted agent is monoclonal antibodies that have displayed promising results in tumors with high HGF/c-MET levels. Rilotumumab (AMG102) is an anti-HGF monoclonal antibody that interferes with c-MET's activation by HGF [Giordano, 2009]. Rilotumumab is currently evaluated in clinical phase I/II studies alone or in combination with the EGFR-blocking antibody panitumumab [Amgen, 2008]. Previous studies have shown that rilotumumab decreases c-MET phosphorylation and can stabilize the progression of certain solid tumors [Wen et al. 2011; Gordon et al. 2010].
MetMAb (OA-5D5), a human, monovalent antagonistic anti-MET antibody [Jin et al. 2008], in preclinical studies was able to inhibit glioblastoma U87 and pancreatic BxPC3 and KP4 tumor xenografts followed by a decrease in cellular proliferation and motility [Jin et al. 2008; Martens et al. 2006]. A recent phase II clinical trial using MetMAb in combination with erlotinib to treat patients with NSCLC resulted in a doubling of patient survival from 6.4 to 12.4 months [Spigel et al. 2010; Zhou et al. 2010]. In this trial, the authors point out that ‘c-MET diagnostic negative tumors’ when treated with MetMAb and erlotinib had a worse overall survival when compared with the erlotinib plus placebo arm [hazard ratio (HR) = 2.52), pointing out that only c-MET-diagnostic positive tumors benefited from the combinational treatment (HR = 0.56) [Khachatryan et al. 2010]. So far, monoclonal antibodies in preclinical and clinical studies have only demonstrated partial or complete response in patients (or cell lines) with high c-MET levels or an HGF/c-MET autocrine loop [Wen et al. 2011; Gordon et al. 2010; Khachatryan et al. 2010; Jin et al. 2008; Vigna et al. 2008; Martens et al. 2006].
In recent years, the therapeutic aim of finding drugs that selectively target a molecule expressed on neoplastic cells has shifted to finding drugs or combinations of drugs that are able to inhibit multiple pathways both in cancer cells and cells of the microenvironment [Sierra et al. 2010]. We have described multiple clinical trials in which c-MET-specific inhibitors are being combined with other RTK inhibitors. This rationale has gained momentum after clinical experience shows that patients who undergo a single-targeted therapy develop drug resistance and relapse. In addition, we have become more knowledgeable with regard to the notion that the tumor microenvironment plays an important role in maintaining the tumor niche; therefore, combination therapies attempt to inhibit neoplastic cells, the vessels and supporting cells (cancer-associated fibroblasts or tumor-associated macrophages) that transport and/or provide nutrients and growth factors to them [Petrelli and Valabrega, 2009]. Finally, the use of multikinase inhibitors has the potential of delaying the development of resistance, since it is known that neoplastic cells are able to undergo an ‘oncogenic switch’ by which the cell that was originally dependant on a single oncogene can rely on the activation of alternative(s) oncogenes [Cepero et al. 2010b; Sierra et al. 2010].
Blocking HGF or c-MET antibodies and TKI appears promising and it is anticipated that many studies will be initiated in the years to come, since c-MET is highly expressed in a wide variety of tumors. All of the c-MET-targeted therapies previously discussed exhibit the potential to enter a phase III study either alone or in combination with other kinase inhibitors and reach the approval of the US Food and Drug Administration in the future.
Potential resistant factors to c-MET inhibitors
As with other TKI inhibitors, sensitive cells/tumors treated with c-MET inhibitors develop resistance [Cepero et al. 2010b; Sierra et al. 2010]. Since no clinical specimens are still available, only preclinical in vitro systems have been able to predict the possible mechanisms of resistance that patients will develop when exposed to anti-MET treatments. So far, three mechanisms of resistance to c-MET inhibitors have been described. First, cells treated with c-MET TKI at high fixed doses develop a dependency on EGFRs [Corso et al. 2010; McDermott et al. 2010]. Both reports conclude that cells harboring high MET copy number will undergo an oncogenic switch that will create an ERBB tyrosine kinase dependency, similar to the oncogenic switch from EGFR to c-MET in NSCLC cells. The second known mechanism of resistance reported by Cepero et al. [2010a] is that when c-MET-dependent NSCLC and gastric cell lines are exposed to increasing doses of c-MET inhibitors amplify wild-type MET and KRAS, it enables cells to overcome the inhibitory threshold of the compound and still sustain high MAPK and PI3K/AKT activity. The third reported mechanism of resistance is the acquisition of a point mutation in the activation loop of c-MET (Y1230H) [Qi et al. 2011]. This mutation had previously been described as a somatic mutation in hereditary and sporadic renal carcinomas [Giordano et al. 2000]; here it has been shown to overcome the inhibitory effect of any c-MET kinase inhibitor.
Summary
With just over 25 years since its first discovery, the c-MET receptor is emerging as an important target for personalized cancer therapy. Inhibition of c-MET receptor activity in vivo has shown promising results in inhibition of tumor cell growth and in overcoming resistance to anti-EGFR therapy, which has now becoming a standard therapy for patients with advanced NSCLC. Results from early phase clinical trials are starting to demonstrate the importance of c-MET/HGF signaling in cancer biology. The challenge will be to explore and discover further other important crosstalk mechanisms involving this pathway, which could lead to further improvement in the efficacy of novel anticancer therapies and improve patient survival.
Footnotes
Acknowledgements
Matthew Joynson, a medical writer, assisted with the styling of this manuscript. The authors wrote and revised the main draft of the article.
Funding
This manuscript was partially supported by the Ontario Research Fund Research Excellence Award (RE-03-020) from the Ontario Ministry of Research and Innovation, Canadian Institutes of Health Research (grant number MOP-64345) and in part by the Ontario Ministry of Health and Long Term Care. MST is the M. Qasim Choksi Chair in Lung Cancer Translational Research. Editorial assistance was supported by Daiichi Sankyo Europe GmbH.
Conflicts of interest statement
Dr J. Rafael Sierra declares no conflict of interest. Dr Ming-Sound Tsao has received honoraria from Daiichi Sankyo Europe GmbH for speaking at scientific symposia.