
Abstract
Renal cell carcinoma (RCC) is the most common type of kidney cancer and is divided into two distinct subtypes, clear cell renal cell carcinoma (ccRCC) and non-clear cell renal cell carcinoma (nccRCC). Although many treatments exist for RCC, these are largely based on clinical trials performed in ccRCC and there are limited studies on the management of nccRCC. Non-clear cell RCC consists of multiple histological subtypes: papillary, chromophobe, translocation, medullary, collecting duct, unclassified, and other rare histologies. Due to variations in pathogenesis and therapeutic response, therapy should be tailored to specific variant histologies. For patients with localized nccRCC, surgical resection remains the gold standard. In the metastatic setting, the standard of care has yet to be clearly defined, and most guidelines recommend clinical trial participation. General therapeutic options include immunotherapy, either as monotherapy or in combination, targeted therapies such as vascular endothelial growth factor tyrosine kinase inhibitors and MET inhibitors, and chemotherapy in certain subtypes. Here we present a review of the incidence and pathogenesis of the various subtypes, as well as available clinical data to support therapeutic recommendations for these subtypes. We also highlight currently available clinical trials in nccRCC and future directions in investigating novel treatment modalities tailored to patients with variant histology.
Introduction
Renal cell carcinoma (RCC) is the most common type of kidney cancer and is one of the most prevalent malignancies in the world. 1 RCC is divided into two distinct subtypes, clear cell renal cell carcinoma (ccRCC) and non-clear cell renal cell carcinoma (nccRCC). Many treatments exist for RCC; however, these are largely based on clinical trials performed in ccRCC. nccRCC consists of multiple histological subtypes, all with varying prognoses and efficacy to therapeutic agents. 2 Therefore, there is a great need to investigate specific treatment modalities tailored to patients with variant histology RCC. Herein, we review the histology and incidence of nccRCC, the pathogenesis of different subtypes, and the available clinical data to support therapeutic recommendations for these subtypes. We also highlight current clinical trials available for nccRCC and future directions for targeting unique biological mechanisms in an ever-evolving treatment landscape.
Histology and incidence
As one of the most prevalent cancer types in the United States, kidney cancer is a major malignancy, comprising an estimated 5% and 3% of all new cancer cases in the United States in males and females, respectively. 3 Among cancers of the kidney, RCC is the most common type, making up over 90% of kidney cancer cases. RCC can further be divided into two subgroups, with the majority being ccRCC (75%), and the remaining being nccRCC (25%).2,4 RCC occurs in the renal tubular epithelium, and each RCC histologic subtype is associated with a distinct pathogenesis.
nccRCC comprises a unique group of malignancies that can further be subdivided into the following histologic subtypes: papillary, oncocytic and chromophobe, collecting duct, molecularly defined renal carcinomas (which include SMARCB1-deficient medullary RCC, translocation, hereditary RCCs), and other renal tumors.5,6 Papillary RCCs (pRCC), which make up 10–15% of all RCCs, arise from a nephron’s proximal and distal convoluted tubules. 6 Historically, pRCC has been divided into Type 1 and Type 2; however, the World Health Organization 2022 classification eliminated the Type 1/2 pRCC subcategorization, given the recognition of frequent mixed tumor phenotypes and different molecular underpinnings across the subtypes. 7 The next most frequent variant histology is chromophobe RCC (chRCC) which accounts for 5–7% of all RCCs and originates from the distal nephron, in contrast to ccRCC which arises from the proximal nephron. 8 Collecting duct RCC (cdRCC) occurs in the distal collecting duct epithelium and represents 1–2% of RCCs. 9 Finally, unclassified or other renal tumors comprise approximately 2–6% of RCCs. 10 Other rare RCC subtypes represent <1% of all RCC tumors, including SMARCB1-deficient medullary RCC, which arises from the calyceal epithelium, and molecularly defined RCC variants, such as TFE3-rearranged, transcription factor EB (TFEB)-rearranged, TFEB-amplified, FH-deficient, succinate dehydrogenase (SDH)-deficient, anaplastic lymphoma kinase (ALK)-rearranged, ELOC-mutated, and other SMARCB1 (INI1)-deficient RCCs. 11 These molecularly defined entities reflect genotype–phenotype relationships. 12 The histology and incidence of the various RCC subtypes are summarized in Table 1. Below we further explore the varying pathogenesis of different RCC subtypes.
Histology and incidence of RCC.

Not a distinct subtype.
RCC, renal cell carcinoma.
Pathogenesis
ccRCC
ccRCC is the most common type of RCC and originates from epithelial cells of the proximal tubule, although some evidence suggests Bowman’s capsule as the site of origin. 13 Chromothripsis is the most common event preceding the clinical presentation of ccRCC and marks the beginning of its pathogenesis. This typically consists of an additional copy of chromosome 5q and the loss of a copy of chromosome 3p, on which the Von Hippel-Lindau (VHL) gene is located.14,15 The VHL gene encodes the VHL tumor suppressor protein (pVHL), which functions as a multi-protein ubiquitin ligase responsible for hypoxia-inducible factor (HIF) poly-ubiquitination and degradation under normoxic conditions. Loss of function of pVHL leads to the accumulation of HIF, resulting in increased expression of genes that control cell proliferation, invasion, and angiogenesis, contributing to the pathogenesis of ccRCC.
Also present in close proximity to the VHL gene on chromosome 3p are tumor suppressor genes Polybromo 1 (PBRM1), a regulator of interferon-stimulated gene factor and a subunit of a SWI/SNF chromatin remodeler complex, and BRCA1-associated protein 1 (BAP1), a ubiquitin carboxyl-terminal hydrolase, which is responsible for modulating gene transcription, DNA damage repair, apoptosis, cellular differentiation, and acts as a deubiquitinating enzyme.16,17 PBRM1 alterations are present in about 40% of patients with ccRCC and have been associated with favorable prognosis, while BAP1 alterations, which occur in approximately 10% of ccRCC tumors, have been associated with poor prognosis.17–19 SETD2 mutations, observed in approximately 10% of ccRCC cases, are also identified as assisting in tumor growth. 20 SETD2 is involved in RNA splicing, DNA methylation, and DNA double-stranded break repair, supporting evidence that loss of SETD2 has a large impact on a plethora of biological processes. 20 VHL, PBRM1, BAP1, and SETD2 are the most common gene alterations in ccRCC. 21
nccRCC
Papillary renal cell carcinoma (pRCC)
In comparison to ccRCC, nccRCC pathologies differ significantly with respect to each histological subtype. Similar to ccRCC, pRCC is predominantly multifocal and bilateral and has a diverse morphology with significantly higher mutation rates overall.15,22
pRCC was previously classified as either Type 1 or Type 2 and most data examining molecular classification is within the context of those labels. Type 1 tumors were characterized by papillae with small ovular nuclei, and molecular alterations consisted of trisomy of chromosomes 7 and 17, and to a lesser extent mutations in chromosomes 3q, 8, 12, 16, 20, or Y.7,23,24 Chromosome 7 contains the MET proto-oncogene on its long arm, and its activation prevents apoptosis, leading to oncogenesis.3,22 Of patients with Type 1 pRCC, 81% were identified to have MET/chromosome 7 alterations and increased MET expression.3,25 In addition to MET mutations, TERT, CDKN2A, CDKN2B, and EGFR were commonly altered in pRCC.3,25
Tumors historically categorized as Type 2 showed inconsistency in driver mutations across tumor types. Observed alterations included mutations in fumarate hydratase (FH), TFE3 fusions, increased expression of NRF2-antioxidant response element (ARE) pathway, CDKN2A silencing, and alterations in CDKN2B, SETD2, BAP1, PBRM1, and TERT.15,23,25,26 Furthermore, mutations in the CpG island methylator phenotype have been reported, leading to inhibited expression of tumor suppressor genes. 3 Type 2 pRCC had also been associated with increased DNA hypermethylation with low overall mutation rates. 25 Type 2 tumors show loss of chromosomes 1p and 9p, and gains of 12, 16, and 20.7,23,21
Type 1 and 2 classifications are no longer used due to mixed tumor phenotypes and molecular diversity, and therefore classification cannot be encompassed by a single well-defined entity. 5 For example, Type 2 pRCC classification has shifted to individual subgroups with different molecular backgrounds, such as tubulocystic RCC, eosinophilic solid and cystic RCC, clear cell pRCC, FH-deficient RCC, SMARCB1-deficient RCC, and MiTF family RCC. Type 1 pRCC is now often regarded as the classical pRCC morphology; however, additional variants with papillary morphology have started to emerge, including papillary renal neoplasm with reversed polarity, thyroid-like follicular RCC, and biphasic hyalinising psammomatous RCC. 5 In addition, while not a WHO classification, pRCC can be divided into molecular subgroups MET-driven and MET-independent. 23
Oncocytic and chromophobe renal tumors
This classification includes oncocytoma, chRCC, and a new category ‘other oncocytic tumors of the kidney’. 5 This new category includes oncocytic renal neoplasm with low malignant potential not otherwise specified for tumors with features in between chRCC and an oncocytoma. It also includes emerging entities such as low-grade oncocytic tumors and eosinophilic vacuolated tumors. 5
There is currently a lack of exact criteria to clearly define subtypes of chRCC, given inconsistencies in distribution between different studies and molecular variations between groups. 5 However, although not an official WHO classification, chRCC has been seen divided into classic variant and eosinophilic variant.27,28 The classic variant is composed of pale cells, while the eosinophilic variant is associated with smaller cells with granular eosinophilic cytoplasm. Typical chRCC is composed of both subtypes in varying proportions. In comparison to ccRCC, chRCC has a threefold lower somatic mutation rate and low immune expression.29,30 Furthermore, patients usually present earlier in the disease course and less aggressively, and the 5-year overall survival (OS) rate among chRCC patients is >80%.15,31,32
chRCC can also be characterized by mitochondrial gene expression and chromosomal aneuploidy, which distinguishes it from other kidney cancers. Chromosomes most commonly lost include 1, 2, 6, 10, 13, and 17; although 3, 5, 8, 9 11, and 18 are also lost at a high frequency.25,31 Mitochondrial gene mutations occur most commonly in TP53 and PTEN, which are tumor suppressor genes that regulate rapid uncontrolled cell division, and less commonly in MTOR, NRAS, TSC1, and TSC2. 28 In addition, mutations of TP53 and loss of transcription factor HNF1B gene are associated with increased aneuploidy and chromosomal instability, giving rise to abnormal cell growth and tumor formation, and consequently a more distinct chromophobe presentation. 33 Furthermore, possibly playing a role in oncogenesis in chRCC are structural changes in the TERT gene promoter, which regulates elongation via telomerase, resulting in short telomeres and kataegis (a pattern of localized hypermutations), ultimately driving oncogenesis.15,30,34
Molecularly defined renal carcinomas
This new subtype under the WHO 2022 classification system includes molecularly defined RCC variants, such as TFE3-rearranged, TFEB-rearranged, TFEB-amplified, FH-deficient, SDH-deficient, ALK-rearranged, ELOC-mutated, and other SMARCB1 (INI1)-deficient RCCs. 5 Below is a closer look at the pathogenesis of various translocation RCCs, SMARCB1-deficient medullary RCC, and hereditary RCC syndromes.
Translocation renal cell carcinoma (tRCC)
This subtype was first recognized in the WHO 2004 renal tumors classification and is now included in the WHO 2022 classification under the ‘molecularly defined renal carcinomas’ group. 5 Similar to pRCC and ccRCC, tRCC originates in epithelial cells of the proximal tubule and is more commonly found in females and younger patients.15,35,36 Chromosomal translocation is the driving event of tRCC and takes place most frequently on chromosome Xp11.2, on which the microphthalmia-associated transcription (MiT) family is located.15,37 MiT genes (TFE3, TFEB, TFEC, MiTF) contribute to cell differentiation, and overactivation of MiTF has been linked to hereditary RCC. TFE3 and TFEB fused to other genes have been present in cases of tRCC.37,38 Gene fusion, specifically with regard to TFE, is generally caused by disordered TFE protein activity due to promoter substitution. Fusion partners and breakpoints vary, but studies have shown distinct partners between TFE3 (ASPSCR1, SFPQ, PRCC, and NONO), TFEB, and MiTF, as well as certain chromosomes (1, 17, and X) that demonstrate the ability to have multiple MiT/TFE fusion partners. 38 Although TFE3 and TFEB are regulators of signaling pathways connected to cancer, the mechanism of how altered TFE protein activity leads to carcinogenesis is unclear. 37 These genes were previously classified together as an ‘MIT family of RCCs’ but are now separated into two distinct types, TFE3-rearranged RCC and TFEB-altered RCC, under the broader subgroup ‘molecularly defined renal carcinomas’. 5
In addition to the translocations, tRCC may also be marked by chromosome 3p loss, as seen in ccRCC with trisomy of chromosome 7 or 17, and in pRCC by 9p21.3 deletions.37,38 Due to its diverse presentation and ability to present similarly to other RCC histologies, tRCC cases may be underestimated or mistaken as a separate histology. 38
SMARCB1-deficient medullary RCC
SMARCB1 (INI1)-deficient renal medullary carcinoma (RMC), formally known as medullary RCC (mRCC), first develops in the calyceal epithelium of the distal nephron, unlike other RCC. 15 The hallmark of mRCC is the loss of SMARCB1, a tumor suppressor gene. Therefore, according to the new 2022 WHO classification, these neoplasms are named as SMARCB1-deficient medullary RCC. 5 RMC is a highly aggressive form of RCC and occurs almost exclusively in younger adults with sickle cell trait.15,25,39
Research suggests that thick ascending limb cells transform into RMC cells associated with ferroptosis resistance programs, allowing survival under conditions favorable to SMARCB1 loss. Ferroptotic cell death is iron-dependent and ferroptosis resistance is associated with high iron concentrations present with the sickle cell trait.40,41
Hereditary RCC syndromes
While the majority of RCCs are sporadic in nature, approximately 3% of RCC cases are inherited and referred to as familial RCC. 34 Although inheritance is generally autosomal dominant across these familial syndromes, having the mutation does not always indicate a family history of RCC, as a proportion of those with the mutation may not exhibit symptoms (incomplete/non-penetrance). The onset of RCC at an early age and the presence of bilateral tumors may indicate hereditary RCC, even in cases without a positive family history. 42
As illustrated in Table 2, among the syndromes most commonly inherited are VHL disease, Birt-Hogg-Dubé (BHD) syndrome, FH-deficient RCC, succinate dehydrogenase-related RCC (SDHRCC), hereditary papillary RCC (HPRCC), hereditary BAP1-associated RCC, and more rarely Cowden/PTEN hamartoma syndrome, tuberose sclerosis, and paraganglioma/pheochromocytoma (PGL/PCC).11,42 Many of these subtypes are now included in the WHO 2022 classification under its entity within the section on molecularly defined renal carcinomas. 5
Hereditary RCC.
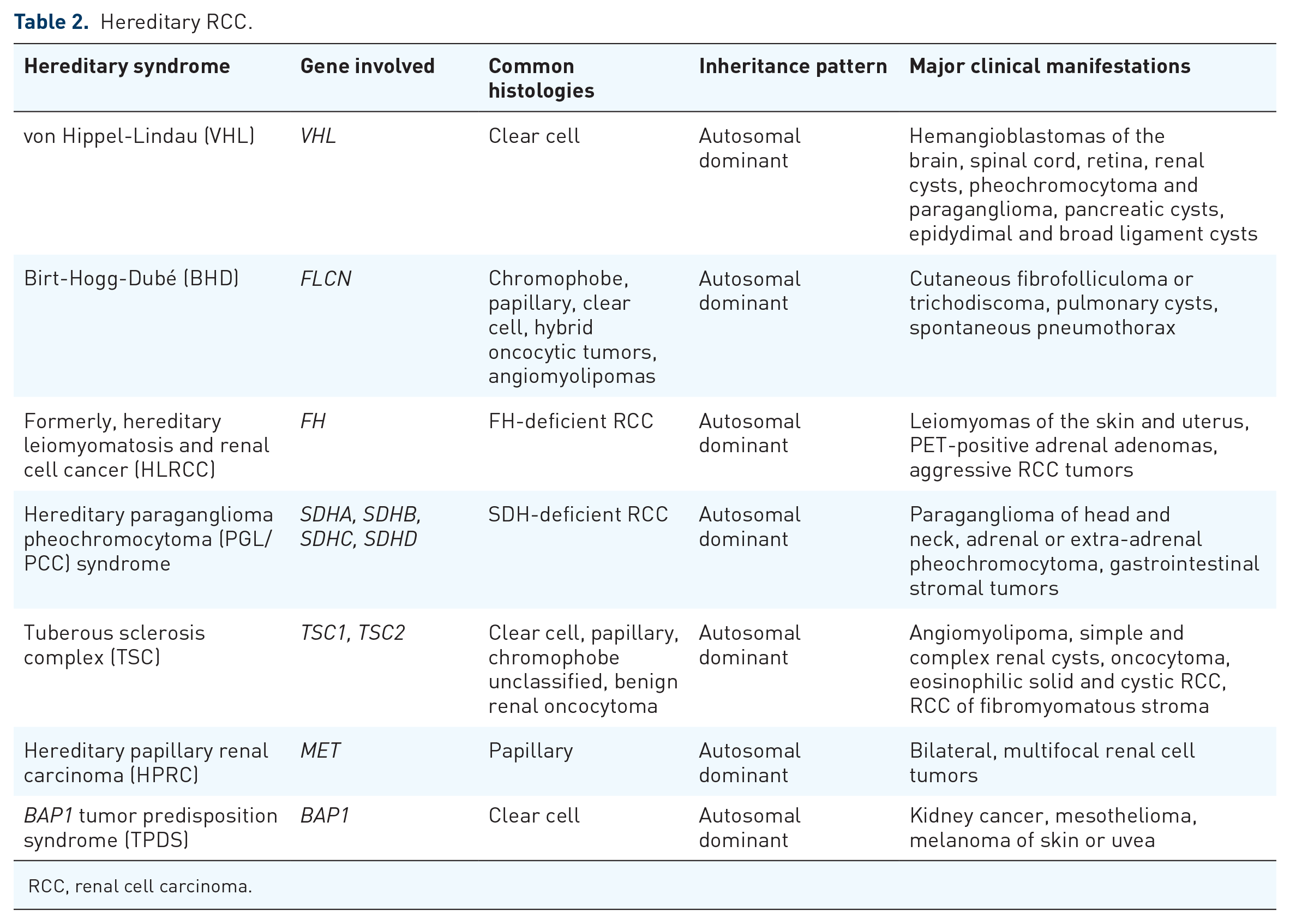
RCC, renal cell carcinoma.
VHL syndrome is an autosomal dominant syndrome characterized by the presence of malignant and benign tumors, manifesting as RCC, hemangioblastomas, and pancreatic cysts. 43 Most cases are familial (80% have an affected parent), though about 20% are de novo. Nearly all patients will express the syndrome by the age of 65. VHL syndrome has an incidence of 1 in 36,000, with two-thirds of patients developing RCC. The majority of VHL patients show a mutation or loss in the VHL gene, which participates in the regulation of HIF and acts as a tumor suppressor.42,43
BHD syndrome is caused by FLCN gene germline mutations, and FLCN inactivation causes mTOR pathway activation, which plays a role in HIF regulation. FLCN mutations are correlated with an increased risk of early-onset ccRCC, chRCC, pRCC, and oncocytoma.34,44 Of the BHD patients, a hybrid oncocytic/chromophobe tumor is the most common.
FH-deficient RCC, formerly known as Hereditary Leiomyomatosis RCC, is mostly seen in cases of papillary RCC, specifically in the formerly categorized Type 2 pRCC. 23 Fumarate hydrase-deficient renal cell carcinoma (FHRCC) is characterized by FH gene inactivation, hindering the Krebs cycle and leading to accumulation of HIF, increased transcription of vascular endothelial growth factor (VEGF), and increased metastatic potential. 7 Also associated with pRCC, specifically Type 1, is HPRCC, characterized by germline MET-activating alterations that are passed down in an autosomal dominant pattern, with incomplete penetrance also presenting in a small portion of sporadic pRCC cases.3,6,7,11,23
SDHRCC is an extremely rare RCC subtype and presents in 0.05–0.2% of cases. 45 It is associated with mutation of the SDHA, SDHB, SDHC, and SDHD genes, which encode SDH, an enzyme vital for cellular metabolism and responsible for converting succinate to fumarate in the Krebs cycle. The most commonly mutated SDHB gene leads to the deficiency of SDH, which, in turn, increases the risk of PGL/PCC. 45
CdRCC
Arising from distal collecting duct principal cells, cdRCC, formerly known as Bellini duct carcinoma, is linked to mutations of SETD2, SMARCB1, homozygous deletion of CDKN2A gene, and alterations in neurofibromatosis Type 2 (NF2), resulting in the loss of a tumor suppressor protein called ‘merlin’ (encoded by NF2).25,46–48 While mutations in NF2 may lead to tumorigenesis, complete loss of the gene can cause NF2 syndrome, and that loss of heterozygosity is associated with poor prognosis and early tumor onset. 47
Alterations and deletions of chromosomes 1q, 8p, and 13q have also been reported in cdRCC pathogenesis. 25 Mutations in the MLL gene are present in about 50% of cdRCC cases, and overexpression of SLC7A1, an oncogenic protein that suppresses ferroptosis, has also been recorded. 48 Generally, cdRCC cases fail to have consistent molecular alterations, contributing to poor prognosis and treatment resistance. 49 Since these tumors are not driven by angiogenesis and do not respond to tyrosine kinase inhibitor (TKI) therapy, they tend to be treated similarly to urinary tract tumors as opposed to parenchymal RCC tumors. 49
sRCC
Rather than being its histological subtype of RCC, sRCC indicates the development of sarcomatoid features in RCC tumors of any histology as a result of dedifferentiation.15,23,50 sRCC presents as malignant spindle-like cells without distinguishable epithelial components. Regions may be homogeneous, uniformly resembling fibrosarcoma or malignant fibrous histiocytoma, or heterogeneous within RCCs.15,51,52 Epithelial regions are often not described as sarcomatoid but instead represent cells of RCC histologic subtypes, most commonly ccRCC in over 80% of sRCC cases. 52 Sarcomatoid features are also present in chRCC and pRCC, and 2–10% of sRCC cases are found in unclassified RCCs. 52 Due to variations in histological subtypes, mutational drivers in sRCC are not well defined.15,51,52
Sarcomatoid dedifferentiation may occur as a result of epithelial–mesenchymal transition (EMT), in which epithelial cells lose their epithelial traits and develop mesenchymal sarcomatoid traits. 51 This occurs when the transcription factors regulating the EMT process cause upregulation of mesenchymal markers and downregulation of epithelial markers. EMT can also contribute to carcinogenesis, possibly explaining sRCC’s aggressiveness, due to the increased metastasis ability accompanying mesenchymal development. 51
Losses on chromosomes 13q and 4q have been reported in sRCC tumors while gains at chromosomes 1, 2, 6, 10, and 17 are common in dedifferentiated chRCCs. 52 TP53 gene expression and its influence on sRCC remain unclear (some studies show under-expression while others show overexpression).15,52–54 BAP1 alterations, CDKN2A deletions, and increased MYC expression have been noted in sRCC cases compared to other RCCs.15,23,50,51 Integrated molecular studies have demonstrated that sRCC exhibits an immune-inflamed phenotype characterized by immune activation, increased cytotoxic immune infiltration, upregulation of antigen presentation machinery genes, and programmed death-ligand 1 (PD-L1) expression, thus increasing sensitivity to immune checkpoint inhibitors (ICIs). 51
Unclassified
Despite the recognition of many RCC histologies, certain subtypes remain undefined. While they may share similarities in morphology and/or immunohistochemistry with the already distinguished histologies, differences prevent them from being grouped within those classifications, and therefore are accordingly labeled as ‘unclassified’.10,55 ‘Unclassified’ is a heterogeneous umbrella term for all tumors that may not fit into any category, or that could fit into multiple, especially as more morphologic similarities between subtypes are being discovered. For example, without the sickle cell trait, a tumor identical in morphology to an mRCC tumor would be categorized as ‘unclassified renal cell carcinoma with renal medullary phenotype’. 39
Treatment
General therapeutic approaches in nccRCC
Data on the optimized treatment of nccRCCs are limited, and treatment recommendations often mirror the approach in ccRCC.56,57 Each subtype has its characteristic molecular profile and signaling pathways that can be used to develop targeted treatments. 57 In localized nccRCC, surgical resection remains the gold standard. 56 While studies have examined VEGF TKI and immunotherapy in the adjuvant setting post-resection for patients with ccRCC, there have been limited adjuvant studies that have included patients with nccRCC. Currently, post-resection the standard of care is expectant monitoring without the use of adjuvant therapy for risk reduction. 56
In the metastatic setting, the standard of care has yet to be clearly defined, and most guidelines recommend clinical trial participation for individuals with variant histology. 57 While there is a lack of phase III trials to guide clinical decision-making for patients with nccRCC, a series of phase II trials have guided treatment selection for patients. While nccRCC has divergent pathogenesis and underlying molecular features, clinical trials for nccRCC have historically enrolled all nccRCC subtypes. Although this has facilitated clinical trial accrual, it has limited the capacity to understand the impact of treatments on outcomes for distinct subtypes. In addition, trials were largely based on the paradigms for ccRCC. 57 Historically, in the targeted therapy era, the ASPEN and ESPN trials established VEGF inhibition as a standard option for patients with nccRCC.58,59 The ESPN trial compared the VEGF inhibitor, sunitinib, to the mTOR inhibitor, everolimus, in nccRCC patients. The median OS was 16.2 months (95% CI 14.2–NA) versus 14.9 months (95% CI 8.0–23.4) in patients who received sunitinib versus everolimus, respectively (p = 0.18). 58 In the ASPEN trial, sunitinib demonstrated a significantly improved median progression-free survival (PFS) of 8.3 months compared to 5.6 months with everolimus (p = 0.16, HR 1.41; 80% CI 1.03–1.92). 59 Although the objective response rate (ORR) was under 20% in these trials, the ASPEN and ESPN trials helped establish sunitinib as the standard first-line therapy in nccRCC in the targeted therapy era.
More recently, ICIs, either alone or in combination with VEGF TKI, have populated the treatment landscape for patients with ccRCC and have been investigated for patients with nccRCC (Table 3). 57 ICIs target programmed cell death-1 (PD-1), PD-L1, or the cytotoxic T-lymphocyte-associated antigen. 60 ICI monotherapy in nccRCC was investigated in KEYNOTE-427 with PD-1 inhibitor pembrolizumab. In all patients, the ORR was 26.7%, the median PFS was 4.2 months (95% CI 2.9–5.6), and the median OS was 28.9 months (95% CI 24.3–NR). 61 CheckMate-374 Cohort B investigated nivolumab monotherapy in nccRCC and found an ORR of 13.6% (95% CI 5.2–27.4) and a median PFS of 2.2 months (95% CI 1.8–5.4). 62 Both these studies helped establish the role of single-agent immunotherapy in the treatment of nccRCC (Table 3). CheckMate-920 evaluated the safety and efficacy of a combination of nivolumab plus ipilimumab, with one of the cohorts consisting of nccRCC patients. Results from this specific cohort showed an ORR of 19.6% (95% CI 9.4–33.9), and a median PFS of 3.7 months (95% CI 2.7–4.6), representing encouraging antitumor activity. 63 HCRN GU16-260 Cohort B investigated the efficacy of nivolumab monotherapy in treatment-naïve patients with nccRCC (Part A) and salvage therapy with nivolumab/ipilimumab in nccRCC patients with tumors unresponsive to initial nivolumab monotherapy (Part B). The ORR in Part A was 14.3% (95% CI 4.8–30.3) with a median PFS of 4.0 months (95% CI 2.7–4.3). In Part B, results showed a low ORR of 6% and a median PFS of 2.8 months (95% CI 0.03–18.9); however, the ORR was not dramatically different from the 11.4% in the ccRCC population. 64 UNISoN (ANZUP 1602) also investigated nivolumab/ipilimumab in nccRCC patients refractory to nivolumab monotherapy and found an ORR of 10% and a median PFS of 2.6 months (95% CI: 2.2–3.8), indicating a minority of patients responding to combination ICI therapy after progression on prior ICI therapy. 65
Prospective immunotherapy trials in nccRCC.

Salvage ipilimumab was administered in patients refractory to single-agent nivolumab at 3 months.
Salvage ipilimumab and nivolumab were administered if progressive disease or stable disease was found 48 weeks after initiation.
ICIs, immune checkpoint inhibitors; mOS, median overall survival; mPFS, median progression-free survival; nccRCC, non-clear cell renal cell carcinoma; NE, not evaluable; NR, not reached; ORR, objective response rate.
ICI-targeted therapy combinations have also been explored in nccRCC (Table 4). COSMIC-021 investigated a combination of cabozantinib (TKI) plus atezolizumab (PD-L1 inhibitor) in nccRCC and showed encouraging clinical response, with an ORR of 31% (80% CI 20–44), and a median PFS of 9.5 months (95% CI 6.4–18.3) with a 1-year PFS rate of 39%. 66 Another phase II trial showed meaningful clinical efficacy with a combination of atezolizumab and bevacizumab in variant histology nccRCC and ccRCC with sarcomatoid differentiation, with an overall ORR of 33% and a median PFS of 8.3 months (95% CI 5.7–10.9). In this trial, 70% of the patient population consisted of variant histology nccRCC, and ORR specifically in this group was 26%. 67 Another phase II trial showed promising efficacy in nccRCC variants who were treated with a combination of cabozantinib plus nivolumab. Particularly in papillary, unclassified, and translocation-associated histologies, the ORR was 47.5% (95% CI 31.5–63.9), and the median PFS was 12.5 months (95% CI 6.3–16.4). 68 Finally, KEYNOTE-B61 investigated pembrolizumab and lenvatinib as first-line treatment for patients with advanced nccRCC. In all, 147 patients were enrolled from 14 different countries. A confirmed objective response was seen in 49% of patients (95% CI 41–57), including 6% of patients with complete response, showing promising antitumor activity in patients previously untreated for advanced nccRCC. 69
Combination immunotherapy plus targeted therapy trials in nccRCC.

ICIs, immune checkpoint inhibitors; mOS, median overall survival; mPFS, median progression-free survival; nccRCC, non-clear cell renal cell carcinoma; NE, not evaluable; NR, not reached; ORR, objective response rate.
pRCC
pRCC comprises the largest proportion of patients with nccRCC; therefore, studies have attempted to test specific therapeutics for this disease subtype. Given that pRCC has classily been associated with MET alterations, a series of studies have tested MET inhibition for this subtype. 70 Foretinib, one of the first MET inhibitors, demonstrated antitumor activity in patients with advanced pRCC in a phase II trial, with an ORR of 13.5% (95% CI 6.7–23.0), and a median PFS of 9.3 months (95% CI 6.9–12.9). Furthermore, the presence of a germline MET mutation was highly predictive of a response compared to those who did not harbor the mutation (5/10 versus 5/57, respectively). 71 The PAPMET trial was a multicenter phase II study among 147 patients that investigated four drugs, sunitinib, cabozantinib, crizotinib, and savolitinib. Cabozantinib demonstrated a longer median PFS of 9.0 months compared to sunitinib at 5.6 months (95% CI 0.37–0.97, p = 0.019), and a higher response rate (23% versus 4%, p = 0.01). 72 This study established cabozantinib as a preferred therapy for patients with pRCC. In addition, the CREATE trial investigated crizotinib in patients with metastatic papillary RCC Type 1 (PRCC1). In this trial, the ORR was 50% (95% CI 6.8–93.2) in the MET + patients, and 6.3% (95% CI 0.2–30.2) in MET − patients, with a 1-year PFS of 75.0% versus 27.3% in patients with MET + and MET −, respectively. 73 SAVOIR, a phase III randomized clinical trial, compared the efficacy of savolitinib and sunitinib in patients with MET-driven pRCC. Although this trial was closed prematurely which impacted the power to detect significant differences in PFS between the two arms, the median PFS was 7.0 versus 5.6 months (p = 0.31) between the savolitinib versus sunitinib, respectively. In addition, the ORR was 27% (95% CI 13.3–45.5) in the savolitinib group versus 7.0% (95% CI 0.9–24.3) in the sunitinib group. Fewer grade 3 or higher adverse events (AEs) were reported in patients treated with savolitinib (42%) versus sunitinib (81%), and AE-related dose modifications occurred in 10 (30%) of savolitinib patients, compared to 20 (74%) of sunitinib-treated patients. 74 Although data on efficacy were limited, this study showed favorable results toward savolitinib over sunitinib in the MET-driven population and a superior safety profile. Finally, the SWOG S1107 was a randomized multicenter phase II trial that investigated MET inhibitor, tivantinib, alone or in combination with EGFR inhibitor, erlotinib, in patients with pRCC. 75 The median PFS was 2.0 months (95% CI 1.8–3.0) versus 3.9 months (95% CI 1.8–7.3), and OS was 10.3 months (95% CI 7.3–15.7) versus 11.3 months (95% CI 6.7–21.9) in the tivanitinib alone and combination groups, respectively. This study found no clinical activity in patients treated with tivantinib alone or in combination with erlotinib. 75 In summary, these results have established MET inhibition as a promising therapeutic target in pRCC, and the PAPMET study established cabozantinib as a standard of care for these patients. 72
Immunotherapy has also played an important role in pRCC management. In KEYNOTE-427, which investigated PD-1 inhibitor pembrolizumab in nccRCC, 71.5% of patients had pRCC. Within the pRCC cohort, the ORR was 28.8% (95% CI 20.8–37.9), with a median PFS of 5.5 months and a median OS of 31.5 months. 61 CheckMate-374, which investigated nivolumab, included 54.5% of patients with pRCC. Partial response was achieved in 8% of pRCC patients, stable disease occurred in 37.5% of pRCC patients. 64
Finally, combinations of therapy have shown some activity in pRCC. For example, COSMIC-021 studied atezolizumab plus cabozantinib and observed an ORR of 47% in patients with papillary histology. 68 CALYPSO showed high response rates in metastatic pRCC patients treated with a combination of savolitinib and durvalumab, with an ORR of 27%, a median PFS of 4.9 months (95% CI 2.5–10.0), and a median PFS of 12 months (95% CI 2.9–19.4) in the MET-driven population. 76 According to the NCCN guidelines for metastatic nccRCC, a combination of lenvatinib and everolimus is one of the recommended regimens. This was based on an open-label, multicenter, phase II study, where a majority of patients had papillary subtype (64.5%). Within the metastatic pRCC cohort, the ORR was 15% (95% CI 3–38), the median PFS was 9.2 months (95% CI 3.5–NR), and a median OS was 11.7 months (95% CI 8.1–NR). 77
chRCC
There are currently no prospective studies in chRCC and insights regarding how to treat this subtype are largely derived from subset analyses based on outcomes of patients with chRCC enrolled in all-comer nccRCC studies. For example, the ASPEN trial compared everolimus to sunitinib in nccRCC patients of various subtypes. 59 The cohort consisted of 14.8% patients with chromophobe histology, of which the median PFS was 5.5 months (80% CI 3.2–19.7) in chRCC patients treated with sunitinib, compared to 11.4 months (80% CI 5.7–19.4) in patients treated with everolimus, showing more favorable results toward everolimus. 59 On the other hand, the ESPN trial compared sunitinib and everolimus in nccRCC, of which 12/68 (17.6%) of patients were chromophobe subtypes. In the chromophobe population, a median OS was 31.6 months in the sunitinib group compared to 25.1 months in the everolimus group. 58 These results bring to light the variation in clinical response within histologic subtypes and warrant further investigation with larger sample sizes.
TKIs have been shown to have varying activity in chRCC. A phase II multicenter study by Lee et al. was one of the first prospective studies to show meaningful activity with sunitinib in patients with nccRCC. However, only three patients with chRCC were enrolled, of which one had an objective response. Due to the limited number of chRCC patients, PFS and OS could not be determined; however, all three patients with chRCC did show tumor shrinkage, with two continuing treatments at the time of analysis without disease progression. 78 Another phase II trial investigated sunitinib in 57 nccRCC patients, 5 (9%) of which were chromophobe histology. 79 All five patients derived clinical benefit from therapy, with an ORR of 40% and a median PFS of 12.7 months (95% CI 8.5–NA).
The combination of VEGF inhibitors and ICIs has been explored in chRCC and has shown varying efficacy. In the KEYNOTE-427 study, 13% of the patients had chRCC, of which the ORR was 9.5% (95% CI 1.2–30.4). The median PFS was 3.9 months (95% CI 2.6–6.9), and the median OS was 23.5 months (95% CI 9.3–NR). 61 Another phase II trial investigated the efficacy of cabozantinib plus nivolumab in nccRCC patients, of which cohort 2 consisted of seven patients with chromophobe RCC. 69 None of the seven patients with chromophobe RCC in cohort 2 showed an objective response, highlighting the heterogeneity of nccRCC and the need for further studies on immunotherapy-based strategies in chRCC. 69
cdRCC
Currently, no standard treatments exist for cdRCC. Although targeted therapies have been studied in cdRCC, the use of cisplatin–gemcitabine doublet therapy is often practiced, due to its similarities with upper tract urothelial carcinomas. 80 A multicenter prospective study investigated gemcitabine plus cisplatin or carboplatin in 23 patients with metastatic cdRCC. Results showed an ORR of 26% (95% CI 8–44), a median PFS of 7.1 months (95% CI 3.0–11.3), and a median OS of 10.5 months (95% CI 3.8–17.1). 81 Another phase II prospective trial investigated sorafenib combined with chemotherapy gemcitabine and cisplatin, with results showing an ORR of 30.8%, a median PFS of 8.8 months (95% CI 6.7–10.9), and a median OS of 12.5 months (95% CI 9.6–15.4). 82 Currently, the NCCN guidelines recommend chemotherapy as a treatment option for patients with cdRCC.
Published data on the efficacy of targeted therapy are scarce, and often not substantial. However, the recent phase II BONSAI trial demonstrated encouraging efficacy with cabozantinib in patients with cdRCC, with an ORR of 35% (95% CI 16–57) and a median PFS of 4 months (95% CI 3–13), suggesting that this TKI could be a promising therapeutic agent for this subtype. 83 Still, larger-scale studies are warranted to investigate more novel and translation approaches to drug therapy in cdRCC.
FHRCC
There are currently no specific treatment guidelines for FHRCC, and in fact some have been diagnosed and treated as sporadic pRCC. The first prospective trial for FHRCC was a phase II study conducted by the National Institute of Health (NIH) investigating bevacizumab (VEGF inhibitor) and erlotinib (EGFR TKI) in patients with advanced FHRCC (n = 42) or sporadic pRCC (n = 41). The ORR was 64% in the FHRCC cohort and 37% for pRCC. The median PFS was 21.1 months (95% CI 15.6–26.6) in FHRCC and 8.7 months (95% CI 6.4–12.6) in pRCC. 84 This proved to be the first and largest prospective study in FHRCC that provided a basis for TKI-based therapies and led to the recommendation of bevacizumab plus erlotinib for treatment of FHRCC in the NCCN guidelines. 85
sRCC
Several studies have highlighted the activity of immunotherapy in sRCC. A post hoc analysis of the phase III CheckMate-214 trial analyzed the effectiveness of nivolumab/ipilimumab versus sunitinib in patients with sRCC. 86 The ORR was 60.8% in the nivolumab/ipilimumab group compared to 23.1% in the sunitinib group, with a complete response rate of 18.09% versus 3.10%, respectively. The nivolumab/ipilimumab group showed a significantly higher PFS of 26.5 months versus 5.1 months in the sunitinib group (p = 0.009), therefore supporting the use of nivolumab/ipilimumab as first-line therapy for patients with sRCC. 86 Another study performed a meta-analysis of four randomized clinical trials with ICIs, of which 226 patients were treated with ICI combinations and 241 with sunitinib in the control arms. 87 Results showed that ICI-based combinations had a higher PFS and OS compared to sunitinib, and an ORR of 50% compared to 20% in the sunitinib group. This study supports the efficacy of ICI-based combinations for cRCC first-line therapy. 87
Prognostic factors in variant histology RCC
Currently, the prognostic prediction for RCC after nephrectomy is based on the American Joint Committee on Cancers tumor-node-metastasis (TNM) staging system, as well as other pathological and clinical variables, such as Fuhrman nuclear grade, and Eastern Cooperative Oncology Group score.88,89 In localized ccRCC, prognostic models such as UISS, SSIGN, and Leibovich were used to predict OS, cancer-specific survival, and metastasis-free survival, respectively.90–92 In metastatic disease, the two most common prognostic models are the Memorial Sloan Kettering Cancer Center risk model and the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC).93–95 In nccRCC specifically, the IMDC model has been validated in predicting OS and time to treatment failure. 96 Recent investigations however have focused more on a new concept, called ‘immunoscore’ as a prognostic factor in RCC. 97 Immunoscore was first used as a prognostic marker in colorectal cancer, particularly in stage I–III colon cancer patients. 98 Based on the quantification of certain lymphocyte populations, particularly CD3+ and CD8 + T cells, this system allowed for scoring ranging from low immune cell densities (score 0) to high immune cell densities (score 4) in areas of tumor center and invasive margins.98–100
These immunoscores may help translate and stratify which patients would benefit from immunotherapies or combination therapy, and potentially predict a high risk of tumor recurrence.98,100 Recently, immunoscores in combination with TNM stage, Fuhrman grade, and WHO/ISUP 2016 grade have been found to better predict disease-free survival, PFS, and OS in clear cell RCC. 101 When investigating immunoscore in nccRCC cases, however, it did not have a significant effect in predicting disease-free survival or PFS. 102 For OS, the predictive effect of immunoscore was only slightly less than ccRCC. 102 This further emphasized the differing landscapes and tumor biology of nccRCC compared to ccRCC, and why there is a continued need for new prognostic models in RCC.
Future direction
Outcomes for patients with advanced RCC remain poor despite advances in therapy. Although the majority of trials conducted have been in ccRCC, several ongoing trials are available for nccRCC. Many clinical trials are continuing to investigate the synergy of ICIs and VEGF inhibitors. For example, the CANI trial is a single-arm phase II study currently investigating cabozantinib, nivolumab, and ipilimumab in patients with advanced RCC with variant histology [ClinicalTrials.gov identifier: NCT04413123]. Another trial, PAPMET 2, is investigating cabozantinib plus atezolizumab versus cabozantinib alone in patients with metastatic pRCC [ClinicalTrials.gov identifier: NCT05411081]. Similarly, SAMETA is an ongoing trial for locally advanced or metastatic pRCC, investigating savolitinib plus durvalumab versus sunitinib and durvalumab monotherapy [ClinicalTrials.gov identifier: NCT05043090]. Perhaps one of the biggest trials in nccRCC, SUNNIFORECAST is a phase II trial with over 300 patients, comparing nivolumab/ipilimumab to standard of care for first-line treatment of nccRCC. This study recently completed enrollment and results are expected to be published in 2024–2025[ClinicalTrials.gov identifier: NCT03075423].
Additional novel therapies beyond ICIs are also being studied, such as one phase II trial investigating whether the addition of radium-223 dichloride to cabozantinib improves outcomes in patients with advanced RCC with bone metastasis, where 20% of the total sample size will consist of nccRCC [ClinicalTrials.gov identifier: NCT04071223]. STELLAR-304 is a randomized open-label phase III study investigating XL092, a novel receptor TKI, in combination with nivolumab versus sunitinib in patients with unresectable, locally advanced, or metastatic nccRCC [ClinicalTrials.gov identifier: NCT05678673].
Overall, the treatment landscape for RCC is always evolving, and many ongoing trials are evaluating novel agents, combination therapies, treatment responses, safety profiles, and sequences of treatment. Regardless, there is a great need for future clinical trials to identify new driver mutations, predictive biomarkers of response, and refined therapies for different subtypes of RCC, to provide patients with the most effective and least toxic treatment.