
Abstract
Prostate cancer is the most common cancer among men and the second leading cause of cancer-related deaths in men in the United States. The treatment paradigm for prostate cancer has evolved with the emergence of a variety of novel therapies which have improved survival; however, treatment-related toxicities are abundant and durable responses remain rare. Immune checkpoint inhibitors have shown modest activity in a small subset of patients with prostate cancer and have not had an impact on most men with advanced disease. The discovery of prostate-specific membrane antigen (PSMA) and the understanding of its specificity to prostate cancer has identified it as an ideal tumor-associated antigen and has revived the enthusiasm for immunotherapeutics in prostate cancer. T-cell immunotherapy in the form of bispecific T-cell engagers (BiTEs) and chimeric antigen receptor (CAR) T-cell therapy have shown exceptional success in treating various hematologic malignancies, and are now being tested in patients with prostate cancer with drug design centered on various target ligands including not just PSMA, but others as well including six-transmembrane epithelial antigen of the prostate 1 (STEAP1) and prostate stem cell antigen (PSCA). This summative review will focus on the data surrounding PSMA-targeting T-cell therapies. Early clinical studies with both classes of T-cell redirecting therapies have demonstrated antitumor activity; however, there are multiple challenges with this class of agents, including dose-limiting toxicity, ‘on-target, off-tumor’ immune-related toxicity, and difficulty in maintaining sustained immune responses within a complex and overtly immunosuppressive tumor microenvironment. Reflecting on experiences from recent trials has been key toward understanding mechanisms of immune escape and limitations in developing these drugs in prostate cancer. Newer generation BiTE and CAR T-cell constructs, either alone or as part of combination therapy, are currently under investigation with modifications in drug design to overcome these barriers. Ongoing innovation in drug development will likely foster successful implementation of T-cell immunotherapy bringing transformational change to the treatment of prostate cancer.
Plain language summary
There are ongoing developments in therapeutic strategies for the treatment of patients with metastatic castrate-resistant prostate cancer. Many of these developments involve the activation of the immune system to target neoplastic prostate cells and tumors. Conventional immunotherapy modalities such as checkpoint inhibitors did not provide robust response in clinical study to warrant a change to the prostate cancer treatment paradigm. However, we are now seeing various agents in the form of bispecific antibodies and chimeric antigen receptor’s which influence T-cell activity and are leading to interesting and promising pre-clinical and clinical results. This review article highlights the biologic rationale for employment of T-cell redirecting therapies for the treatment of prostate cancer, and reviews much of the exciting data emerging within the field.
Introduction
Prostate cancer (PCa) is the most common malignancy in men with an annual incidence of more than 280,000 new cases within the United States. 1 The incidence of metastatic disease has steadily increased over the years, and advanced disease remains largely incurable. 2 Although androgen deprivation therapy remains the initial treatment modality in the setting of advanced disease, development of treatment resistance conferring a castrate-resistant state is an inevitable reality. We have witnessed stepwise advances in our therapeutic approach to metastatic castration-resistant prostate cancer (mCRPC) with the advent of next-generation androgen receptor signaling inhibitors (ARSIs), cytotoxic chemotherapy, and targeted therapies for a subset of patients. However, these therapies are largely associated with poor tissue selectivity, an abundance of toxicity, and limited durability in responders. 3
With the success seen in other tumor types, there was hope that immune checkpoint blockade (ICB) would offer another therapeutic option for patients with mCRPC. However, it has not had a transformative impact in the treatment of PCa. Large phase III clinical studies evaluating ICB targeting programmed cell death protein 1 (PD-1), programmed death-ligang 1 (PD-L1), and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) have demonstrated limited efficacy and ICB is only approved for patients with metastatic prostatic cancer that have microsatellite instability-high or mismatch repair-deficient status found in their tumor.4–7 PCa harbors an immunologically ‘cold’ tumor microenvironment (TME) with low tumor mutational burden (TMB), thereby limiting neoantigens for immune recognition and lack of development of antitumor immunity. Furthermore, immune cell recruitment and localization within the PCa TME vary across lesions, creating spatial heterogeneity to the immune milieu. 8 T-effector cells are often restricted to tumor-adjacent stroma and benign glandular tissue, whereas the immune infiltrate within clusters of malignant cells exhibits an exhausted T-cell phenotype with a predominance of M2-polarized tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T-cell (Treg) subsets.9,10 This intratumoral immune infiltrate heterogeneity along with immunosuppressive factors is thought to limit robust immunogenic response.
Strategies employing adoptive cellular therapy are actively under investigation using overexpressed self-antigens and may have the ability to reverse or bypass mechanisms of T-cell anergy in PCa. Immune checkpoint inhibitors modulate coinhibitory and costimulatory signaling but have no direct effect on the T-cell receptor (TCR/CD3) or its activation signaling via major histocompatibility complex (MHC) antigen presentation. We are now witnessing a renewed focus on direct T-cell activation as a target for cellular therapies with hopes to leverage the success seen in hematologic malignancies into opportunities for patients with PCa. Early efforts with autologous activated cellular therapy demonstrated proven yet limited success, and Sipuleucel-T holds National Comprehensive Cancer Network (NCCN) guideline category 1 recommendation for use in select patients with mCRPC.11,12 The therapeutic potential of bispecific antibodies (bsAbs) and chimeric antigen receptor (CAR) T-cells is tremendous, and may allow for targeted antigen-dependent T-cell activation and thus more directed antitumor immunity compared with prior immunotherapy modalities. 13 Various antigen targets are under active investigation with rapid drug design integrating prostate-specific membrane antigen (PSMA), six-transmembrane epithelial antigen of the prostate 1 (STEAP1), and KLK2, among other self-antigens. We review the preclinical and early-phase trial data surrounding T-cell redirecting therapies in mCRPC with a focus on therapies engineered to PSMA as the targeted ligand (Figure 1).
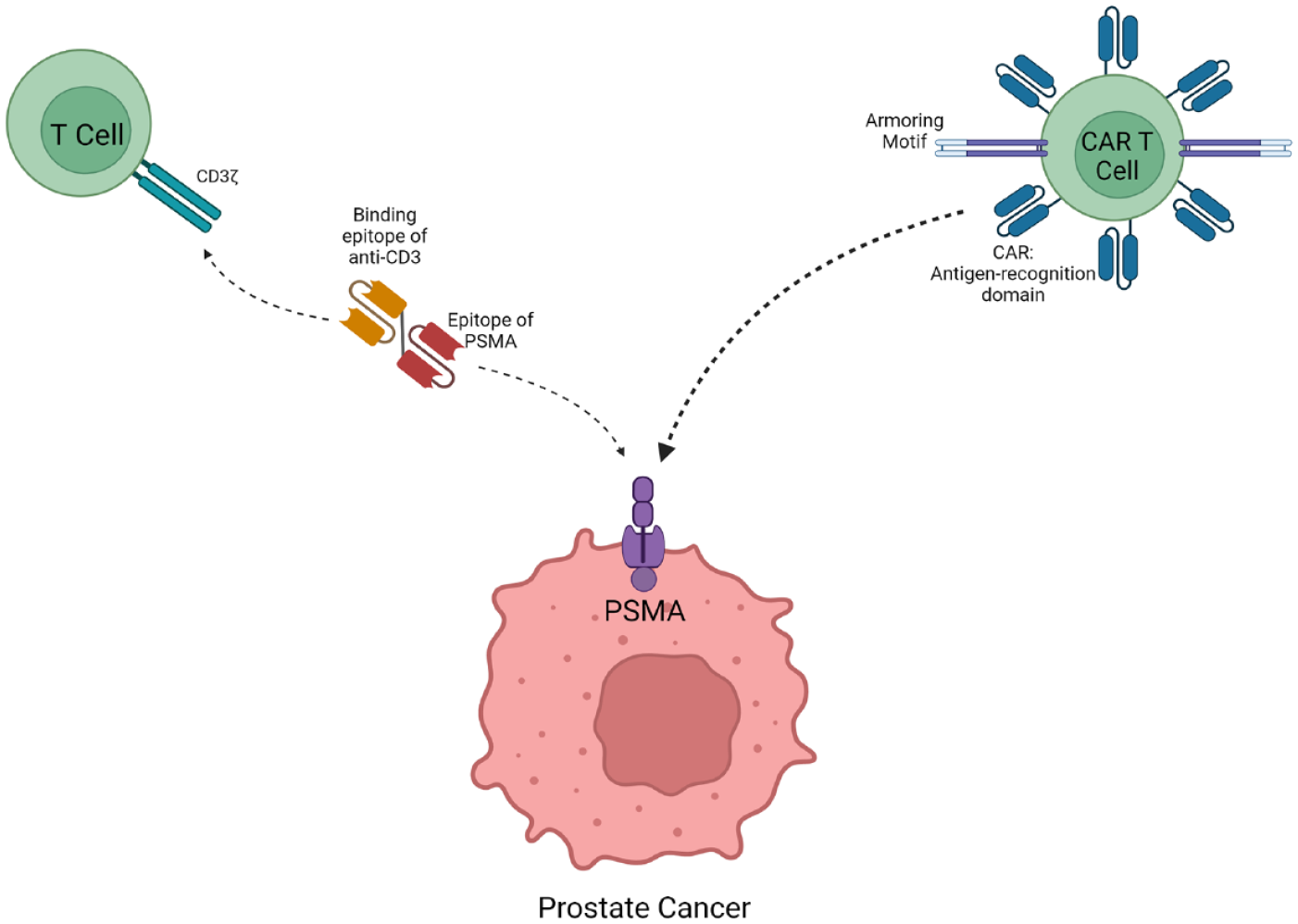
Schematic of the immune synapse between PSMA-expressing prostate cancer cells and T-cell immunotherapeutics (BiTE anitbody on the left and a CAR T-cell on the right).
PSMA as an antigen target
PSMA, also known as folate hydrolase 1, is a type II transmembrane glycoprotein first described in the 1980s during the initial molecular characterization of the LnCAP cell line.
14
Endogenous low-level expression is localized in the salivary gland, proximal renal tubules and duodenum where it regulates folate metabolism,
15
and the central nervous system where its catalytic activity processes N-acetyl-
PSMA is typically localized to the apical surface of epithelial cells in non-neoplastic tissue, where drug delivery is limited, thereby ideally mitigating ‘on-target, off-tumor’ toxicity. 21 Nonetheless, salivary gland hypofunction and xerostomia have been dose-limiting toxicities from PSMA-targeted radionuclide therapy studies, and this off-target effect is a concern. 22 PSMA is not expressed within normal vascular endothelium, but emerging data have demonstrated increased and detectable PSMA expression within aberrant neovascularization in the proximity of various nonprostatic solid tumor malignancies. 23 While this may provide a conduit for PSMA-targeted drug development in other advanced solid tumors, it may also be a source of nonspecific tissue toxicity that requires further investigation.
The diagnostic and therapeutic potential of PSMA as a target ligand is highlighted by the ongoing success of PSMA theranostics and radioligand therapy. 177Lu-PSMA-617 is a novel radioligand therapy which combines a small-molecule inhibitor (PSMA-617) with a β-emitting radionuclide (177Lu). Preliminary studies with 177Lu-PSMA-617 in mCRPC revealed encouraging antitumor activity with biochemical and radiographic responses in patients with advanced disease.24,25 In the landmark phase III VISION trial, 831 patients with mCRPC treated with prior cytotoxic chemotherapy were randomly assigned in a 2:1 ratio to either 177Lu-PSMA-617 plus protocol-permitted standard care or standard care alone. Treatment with 177Lu-PSMA-617 was associated with significantly prolonged progression-free survival (PFS) (median, 8.7 versus 3.4 months), overall survival (OS) (median, 15.3 versus 11.3 months), time to symptomatic skeletal events (median, 11.5 versus 6.8 months), and time to worsening of health-related quality of life and pain (median, 5.7 versus 2.2 months). 26 These results led to the Food and Drug Administration (FDA) approval for 177Lu-PSMA-617 in patients with mCRPC, and there are multiple ongoing studies of novel radioligand targeted therapies in patients in various PCa disease settings, including in castrate-sensitive disease.
Clinical study employing PSMA as a targeted ligand is not limited to radioligand therapy, as antibody drug conjugates (ADCs) are now under investigation employing various cytotoxic payloads, including, but not limited to, the antimicrotubule agents maytansinoid-1 (DM1) (NCT00052000, NCT00070837) and monomethyl aurisatin E (MMAE) (NCT01414283, NCT01695044). The clinical testing and early success of PSMA-targeted radioligand therapies and ADCs demonstrate the promise for integrating PSMA into novel cellular T-cell redirecting therapeutics.
Integrating PSMA into adoptive cellular therapy
PSMA and CAR-T cellular therapy
CAR T-cell therapy’s early success in hematologic malignancies heralded a new era for immunotherapy and was considered a revolution in cancer drug discovery. Since its initial application in patients with relapsed refractory B-cell acute lymphocytic leukemia, six novel CAR-T therapies have gained FDA approval in various hematologic malignancies. However, application of adoptive cellular therapy in solid tumors has not matched the same rapid rate of success. Various barriers, including factors intrinsic to structural elements in CAR design and fitness, as well as complexity of immune microenvironment within solid tumors perhaps explain the more limited progress in CAR T-cell therapies in solid tumor malignancies.
Conventional CAR constructs contain three modular domains: an ectodomain, transmembrane domain, and an endodomain. The ectodomain comprises a synthetic immune receptor containing a single-chain variable fragment (scFv) which recognizes and binds to tumor-associated antigens (TAAs). The TAA binding domain directly connects with the T-cell signaling (CD3ζ) and costimulatory ligands (such as CD28 and/or 4-1BB) within the endodomain for signal transduction leading to sustained T-cell activation and proliferation. Newer generation CAR constructs have incorporated additional costimulatory and pro-inflammatory molecules within the endodomain which have been a successful strategy to further enhance T-cell cytotoxicity.
The process of TAA-mediated T-cell activation is independent of MHC or antigen processing by antigen-presenting cells making this an appealing approach for self-antigen recognition. 27 PCa has the unique ability to downregulate expression of MHC class I or manipulate the TME to prevent MHC epitope interactions as a means of bypassing immunosurveillance.28,29 CAR-T delivers immunologic cytotoxicity directly to TAA-expressing cancer cells, therefore bypassing potential tumor mechanisms of immune escape. Furthermore, PSMA is a glycosylated protein which prohibits TCR recognition and T-cell targeting. CARs have the unique ability to circumvent this immunologic barrier allowing antigen recognition. 30 Together, this supports a strong biologic rationale for the use of PSMA as an optimal TAA to target with CAR therapy in patients with PCa.
Preclinical data
Early preclinical data demonstrated the promising in vitro efficacy and potent immune response triggered by first- and second-generation anti-PSMA CAR-T constructs in various PSMA+ cell lines. Subsequent animal studies employing a first-generation CAR demonstrated complete tumor eradication in a subset of PSMA-expressing orthotropic xenografts, but limited activity in others.31,32 Building on this success, second-generation CAR constructs were created with additional costimulatory domains and demonstrated improved T-cell proliferation and cytokine production in PSMA-expressing tumor mouse models. 33 However, subsequent iterations of CAR constructs with third-generation CARs did not reliably improve efficacy. In fact, third-generation CARs containing costimluatory domains, namely CD28 and 4-1BB, in addition to CD3ζ, led to exhaustion and impaired preclinical T-cell activity. 34 Altogether, the preclinical data surrounding these CAR T-cell models did not provide evidence relating to their ability to overcome immunosuppressive factors within the TME that was sufficient to warrant further clinical study.
One impactful innovation in CAR design was the addition of a dominant-negative transforming growth factor (TGF)-βRII (dnTGF-βRII) domain to PSMA-targeting CAR-T constructs. TGF-β is actively secreted and abundant within the PCa TME and not only suppresses the tumor immune system but also directly promotes neovascularization and metastasis. Effector T-cells endogenously express TGF-β receptors. Activation of the receptor induces intracellular signaling which suppresses cytokine production, reduces cytotoxicity, and inhibits T-cell proliferation in response to antigen stimulation. 35 Early work in cloning the type I TGF-β receptor unveiled a heterodimer binding domain required for kinase function and intracellular signal transduction. 36 Synthetic dominant-negative TGF-βRII peptides which lack the intracellular domain can effectively inhibit TGF-β signaling. 37 These studies provided the biologic rationale for coexpressing dnTGF-βRII within PSMA-specific CAR T-cells in PCa models. Encouragingly, this TGF-β-insensitive CAR construct effectively increased T-cell proliferation and cytokine production and led to long-term eradication of PSMA-expressing tumors in mouse models. Compared with wild-type PSMA CAR T-cells which retain TGF-β signaling, these TGF-β-insensitive CAR T-cells induced a potent and durable immunogenic response resistant to exhaustion. 38
Another promising iteration of PSMA-targeting CAR T-cells in the preclinical setting is a construct which integrates an interleukin 23 (IL-23) monoclonal antibody (mAb) with PSMA-targeting antibody. IL-23 is an inflammatory cytokine which is highly expressed in human tumors and promotes angiogenesis while reducing CD8+ T-cell infiltration and fostering an M2 macrophage cell predominant phenotype. 39 Moreover, MDSCs express IL-23 within the PCa immune milieu contributing to activation of androgen receptor pathway signaling and development of castration-resistant disease. 40 Investigators have created a panel of IL-23mAB-PSMA CARs, including a novel duo-CAR which coexpresses engineered IL-23-specific CAR with PSMA-specific CAR, a single CAR which links IL-23-specific mAb and PSMA-specific mAb, and PSMA CAR with soluble IL-23 mAb which captures secretory IL-23 within the TME. These models demonstrated impressive T-cell expansion when infused in mice, as well as tumor eradication in PC3-PSMA mouse xenografts. 41
Completed and ongoing clinical studies
One of the first CAR T-cell studies in patients with metastatic PCa was a phase I dose-escalating trial assessing safety and tolerance of a second-generation PSMA-targeting CAR. The study enrolled seven patients across three dose levels. At a dose of 1 × 107 CAR+ T-cells/kg, two patients had stable disease for >6 and >16 months, respectively. At a higher dose level, all patients developed high-grade fevers associated with increased levels of IL-4, IL-8, IL-10, sIL-2ra, and IL-6 suggesting T-cell activation. 42 In a subsequent phase I dose-escalation study, five patients were treated with first-generation PSMA CAR T-cells with concurrent continuous infusion of low-dose IL-2 as a means to augment T-cell expansion. Of five patients treated, two achieved clinical partial responses with PSA declines of 50% and 70%, delays in PSA progression of 78 and 150 days, as well as a minor response in a third patient. 43 Although prostate-specific antigen (PSA) responses and engraftment were observed in patients in both studies, the overall limited treatment efficacy in both trials dampened the enthusiasm that CAR T-cells would provide practice-changing advances in PCa as rapidly as they had in select hematologic malignancies.
However, recently reported early-phase trials are providing more encouraging data. Topline results from a highly anticipated open-label, multicenter, 3 + 3 dose-escalating phase I trial employing P-PSMA-101 have been reported. P-PSMA-101 is a CAR engineered via a novel platform using the piggyBac DNA Modification System, which allows for the production of T-cell subsets with a preference for stem cell memory cells that can later lead to improved effector T-cell expansion within the TME. Patients were pretreated with a standard 3-day fludarabine/cyclophosphamide lymphodepletion regimen. At the time of data presentation, the ongoing trial had enrolled 10 heavily pretreated patients, who received an average of 7 prior lines of therapy. One enrolled patient had complications leading to death of unclear etiology. 44 Of the 10 patients, PSA declines of >50% were observed in 3 patients, and >99% in 1 patient. Altogether, seven patients had a PSA decline, and three of the four patients who had pretreatment and post-treatment F-fluorodeoxglucose (FDG) and PSMA-PET imaging demonstrated marked radiographic responses (including complete responses). Correlative studies also noted a concordant decline in circulating tumor cells (CTCs), and post-treatment tumor biopsy demonstrated infiltration by P-PSMA-101 CAR T-cells and a confirmed pathologic complete response. Aside from the early death in a patient case, all treatment-related toxicities were grade 1–3 in severity and were managed effectively. 45 This trial remains active and presentation of data from the full study population is highly anticipated (Table 1).
Ongoing studies evaluating PSMA-targeted CAR T-cell therapy.
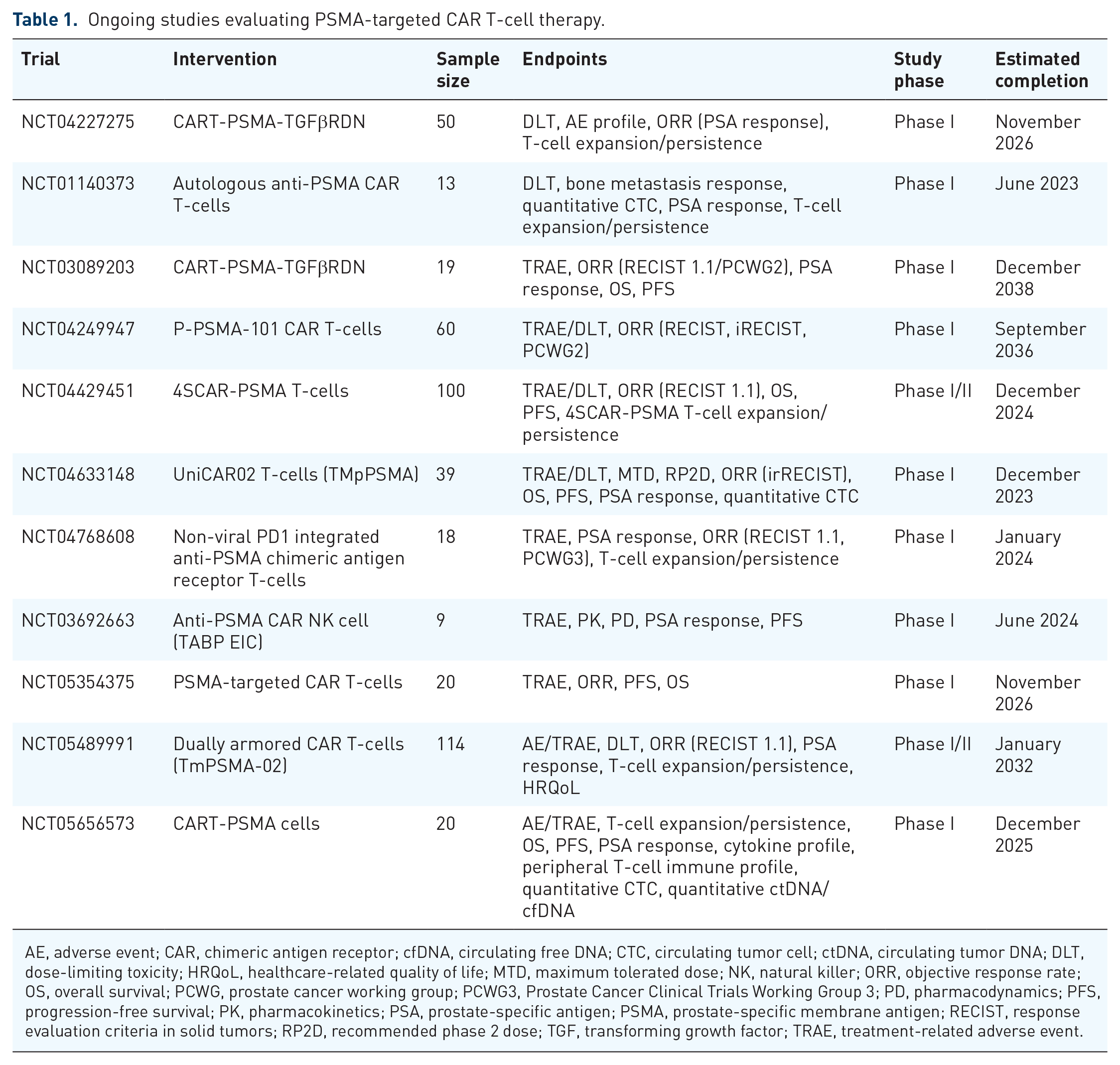
AE, adverse event; CAR, chimeric antigen receptor; cfDNA, circulating free DNA; CTC, circulating tumor cell; ctDNA, circulating tumor DNA; DLT, dose-limiting toxicity; HRQoL, healthcare-related quality of life; MTD, maximum tolerated dose; NK, natural killer; ORR, objective response rate; OS, overall survival; PCWG, prostate cancer working group; PCWG3, Prostate Cancer Clinical Trials Working Group 3; PD, pharmacodynamics; PFS, progression-free survival; PK, pharmacokinetics; PSA, prostate-specific antigen; PSMA, prostate-specific membrane antigen; RECIST, response evaluation criteria in solid tumors; RP2D, recommended phase 2 dose; TGF, transforming growth factor; TRAE, treatment-related adverse event.
Clinical trials evaluating dnTGF-βRII coexpressing PSMA CAR T-cells are now underway. Integration of a dominant-negative TGFβRII moiety provides a colloquial ‘armor’ which may render the CAR construct resistant to TGFβ-mediated immunosuppression. The first-in-human phase I study of TGF-β-resistant PSMA-redirected CAR T-cells demonstrated promising clinical efficacy, safety, and immunogenicity on biologic correlative analyses. The study employed lentiviral transduced PSMA-TGFβRDN autologous CAR T-cells administered with and without cyclophosphamide/fludarabine conditioning in a 3 + 3 dose-escalation design. Thirteen patients received CAR T-cell infusion across four dose levels. A PSA decline of >30% was observed in four patients, including one patient with undetectable levels. Five patients had stable disease at 3-month imaging assessment. The median PFS and median OS were 4.4 and 15.9 months, respectively. Grade ⩾2 cytokine release syndrome (CRS) was observed in five patients. One patient developed grade 4 CRS with concurrent sepsis leading to death (notably, this patient also had a marked clonal CAR T-cell expansion and >98% reduction in PSA). CART-PSMA-TGFβRDN cell expansion occurred in all patients, and cell levels peaked within the first 14 days. Magnitude of expansion generally correlated with the dose level, and patients who received prior conditioning chemotherapy had markedly increased cell concentrations compared with nonconditioned patients. A considerable decline in soluble TGF-β levels was observed in the patient with the highest degree of CART-PSMA-TGFβRDN cell expansion. 46
A follow-up multicenter, open-label, phase 1 trial, CART-PSMA-02, further evaluated the safety and feasibility of CART-PSMA-TGFβRDN T-cells. Topline results included data from nine patients across two cohorts: dose level 1 (1–3 × 107 cells) and dose level 2 (1–3 × 108 cells) with lymphodepletion chemotherapy and anakinra prophylaxis. Clinical activity was demonstrated in four of the five evaluable patients who received ⩾0.9 × 108 cells with radiographic stable disease at day 28, and decreases in serum PSA occurring in four of the seven patients (with >50% decreases observed in two of the five evaluable patients at day 28). 47 Unfortunately, two patients suffered from grade 5 adverse events (one immune-effector cell-associated neurotoxicity syndrome with associated macrophage activation syndrome, and one nonspecific immune-related toxicity). Correlative analysis of serum and peripheral blood samples showed typical patterns of immune-effector response in all patients, but samples from both patients with grade 5 toxicity showed an elevated inflammatory signature with higher levels of IL2, IL6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-18. 48 Given two patient deaths on study during dose escalation, the trial was suspended.
PSMA and bispecific T-cell engager therapies
Bispecific T-cell engager (BiTE) immunotherapy is an alternative approach to T-cell redirection within solid tumor malignancies. This therapeutic class has shown considerable benefit in select hematologic malignancies and BiTE constructs are now under active investigation for metastatic PCa. BiTE immunotherapies are unique antibody constructs which harbor two distinct scFv domains – one epitope with affinity for TAAs and the other with affinity for CD3ζ on T-cells. The BiTE molecule can serve as a physiologic bridge between the tumor cell and the effector T-cell, leading to an immune synapse on the surface of tumor cells. Upon direct engagement of the CD3 costimulatory domain, T-cell-mediated cytolytic activity is directed to the tumor cell independent of TCR/MHC costimulation.
Pasotuxizumab (also known as AMG 212 or BAY 2010112) was one of the first BiTE immunotherapies to enter clinical study for PCa. The anti-PSMA/CD3 BiTE construct demonstrated impressive selectivity for PSMA-expressing target cells, with effective PSMA-dependent activation and cytokine release of T-cells and tumor cell lysis. In PSMA-expressing tumor xenografts in NOD/SCID mice, pasotuxizumab was effective in both delaying tumor growth and inducing rapid tumor shrinkage with complete remission. 49 A subsequent phase I, open-label, dose-escalation study of pasotuxizumab was undertaken in patients with advanced CRPC. Primary objectives of the study were to determine the safety and maximum tolerated dose (MTD) of drug administered by subcutaneous injection or continuous IV infusion (cIV). Secondary endpoints included pharmacokinetics and PSA and radiographic tumor responses. The study enrolled 31 and 16 patients for treatment with subcutaneous and cIV pasotuxizumab, respectively. TRAE ⩾grade 3 was reported by 87% of patients in the subcutaneous (SC) cohort with no treatment-related deaths. However, at interim data review, it was found that all evaluable patients in this cohort developed neutralizing antidrug antibodies (endogenous antibody formation to the idiotope of the drug) and this dosing formulation was deemed nonviable. In the cIV cohort, 81% of patients reported a ⩾grade 3 TRAE. This formulation did not lead to antidrug antibodies, and 14 of the 16 evaluable patients showed a decline in PSA throughout study treatment. PSA response was dose dependent, and two patients had durable responses. On exploratory analysis, there was also a dose-dependent reduction in measurable CTCs with cIV treatment. 50 Although clinical efficacy was observed, the short half-life necessitating cIV administration was cumbersome and impractical limiting further drug development. A decision was made to transition clinical study to a novel half-life extended (HLE) BiTE that may be administered by short intravenous infusion. 51
AMG160 (acapatamab) is a second-generation anti-PSMA/CD3 BiTE. The construct was designed to include an additional Fc fragment linked to the BiTE molecule core which effectively extends its half-life by enabling endogenous transcytosis and recycling mechanisms. 52 The HLE BiTE formulation allows for short-term intravenous infusion every 14 days which is more practical and cost-effective compared with its predecessor compound. Preliminary results from a phase I study assessing safety, tolerability, pharmacokinetics, and antitumor activity have been reported. Of 32 patients treated across six dose levels, an MTD was not reached. Toxicity was predictable and CRS was the most common adverse event occurring in 84.4% of patients and was effectively managed by established supportive measures. A PSA reduction was observed in 63% of patients. 53 Focus is now on a novel BiTE (AMG 340) which harbors a low-affinity anti-CD3 arm as a strategy to reduce off-target immune activation and CRS (Table 2).
Ongoing studies evaluating PSMA-targeted bispecific T-cell engagers.

AE, adverse event; BiTE, bispecific T-cell engagers; cfDNA, circulating free DNA; CTC, circulating tumor cell; ctDNA, circulating tumor DNA; DLT, dose-limiting toxicity; HLE, half-life extended; HRQoL, healthcare-related quality of life; MTD, maximum tolerated dose; ORR, objective response rate; OS, overall survival; PCWG, prostate cancer working group; PD, pharmacodynamics; PFS, progression-free survival; PK, pharmacokinetics; PSMA, prostate-specific membrane antigen; RECIST, response evaluation criteria in solid tumors; RP2D, recommended phase 2 dose; TRAE, treatment-related adverse event; PSA, prostate-specific antigen.
Innovative BiTE constructs are currently in development that will hopefully optimize drug bioavailability, extend half-life, and thereby further improve therapeutic index. APVO414 is an anti-PSMA/CD3 BiTE which was developed through the ADAPTIR™ technology platform. The APVO414-ADAPTIR format entails two scFv homodimers, each bivalently targeting CD3 and PSMA. The unique homodimer structure has been associated with longer half-life, improved stability, and improved potency in preclinical study. 54 Most patients enrolled in the initial clinical study developed antidrug antibodies with high titers (1:250,000) thereby deeming the investigational agent nonviable in its current form. Similar issues with compound immunogenicity and antidrug antibody formation was observed in clinical study of JNJ-081, a BiTE construct created with the novel DuoBody® platform which enables rapid Fab-arm exchange to created bsAbs. Antidrug antibodies and drug clearance were observed in patients treated with the cIV and subcutaneous formulations, leading to premature closure of the trial. 55
HPN424 is a first-in-class trispecific T-cell-activating construct derived from the TriTAC engineering platform. In addition to CD3 and PSMA binding domains, the agent has a third domain which targets albumin to improve drug stability and prolong serum half-life. A phase I/IIa study enrolled 80 patients with mCRPC who had received on average six prior systemic regimens. Patients were dosed across 15 cohorts, and MTD has not been reported to date. All grade CRS occurred in 63% of patients, with only 4% being ⩾grade 3 in severity. A reduction in PSA from baseline was noted in 21% of patients, and a reduction in CTCs was seen in 57% of patients with measurable CTC at baseline. 56 The overall safety, tolerability, and efficacy signals were not sufficient to warrant further study of the compound in its current form (Table 3).
Construct design and limitations of select ongoing and completed studies evaluating PSMA-targeting T-cell immunotherapy.

CAR, chimeric antigen receptor; CRS, cytokine release syndrome; IL-2, interleukin 2; PSMA, prostate-specific membrane antigen; TGF, transforming growth factor.
Perspectives and ongoing work
T-cell redirecting immunotherapies have had transformative impact on various hematologic malignancies, either in the form of CAR T-cell therapy or T-cell engaging bsAbs. Replicating the same biological efficacy in PCa has been a challenge, yet the progress made thus far is far from disappointing as we are likely in the early stages of a potential paradigm shift in this disease space. CAR T-cell and BiTE therapies both face similar barriers that limit efficacy in PCa, which include factors attributed to the TME and also factors inherent to drug design and delivery.
Identification of an optimal tumor antigen with a tumor-restricted expression pattern is necessary to facilitate effective T-cell redirection, and the advent of PSMA as a surface biomarker meets this requirement. 57 We have an incomplete understanding of the endogenous role of PSMA which can be expressed at low levels within various secretary organs and within neovasculature in the proximity of solid tumors. Although PSMA expression within non-neoplastic tissue is at basal low levels, it does provide the risk of ‘on-target off-tumor’ toxicity. However, the recent success of PSMA theranostics serves as a proof-of-principle that baseline off-target expression is not a significant barrier to its therapeutic potential. In fact, PSMA expression within the tumor neovasculature theoretically allows for dual targeting of both the tumor and tumor vasculature allowing for influx of immune cells via the damaged endothelium further leading to antitumor effect. 13 Together, we can consider PSMA as an ideal target for T-cell immunotherapy drug development.
There are alternative TAAs with expression patterns suitable for PCa-targeted therapies. In fact, there is a breadth of ongoing clinical research with T-cell redirecting therapies tailored for prostate stem cell antigen (PSCA) and STEAP1, among others. PSCA is a large glycoprotein expressed on the cell surface of both normal prostate glandular tissue and prostate adenocarcinoma, but with markedly higher expression in malignant cells and expression correlates with Gleason grade and disease aggressiveness. 58 PSCA expression is not limited to prostate tissue, however, and ample expression has been well established on pancreatic, gastric, and bladder tissue. As such, there are ongoing early-phase studies investigating PSCA-targeting BiTE and CAR T-cell studies for PCa as well as basket studies inclusive of a variety of advanced solid tumors. STEAP1 is similarly overexpressed on malignant prostate tissue, as well as a variety of other solid and hematologic malignancies. STEAP1 is a 39.9-kDa protein with six transmembrane motifs expressed at surface cell–cell junctions, with notable expression in prostatic secretory epithelium. 59 STEAP1-targeting T-cell engagers are in development, and highly anticipated early-phase studies are assessing safety and tolerability in patients with PCa.
Perspectives on drug delivery
The role of lymphodepleting conditioning prior to delivery of T-cell redirecting immunotherapy remains unclear. With respect to CAR-T administration, utilization of conditioning chemotherapy has been inconsistent throughout early clinical study in PCa. Conditioning chemotherapy with cyclophosphamide and fludarabine provides well-established effects on tumor immune contexture, ‘depleting’ Tregs which favors activity of antitumor T-cells, as well as facilitating adoptive T-cell proliferation, persistence, and effector function.60,61 Conventional chemotherapy regimens have proven immunologic effects as well, including increasing tumor neoantigens and disrupting immune-suppressive pathways. 62 Narayan et al. 46 demonstrated benefit to preconditioning lymphodepletion in patients with PCa treated with CART-PSMA-TGFβRDN, in which conditioning chemotherapy prior to cell transfer markedly enhanced CAR T-cell expansion compared with cohorts in the same study without conditioning therapy. However, myeloablative chemotherapy poses a significant risk of immunosuppression and infection, which is particularly concerning, given the risk of concurrent CRS. Moving forward, alternative study designs may incorporate low-dose radiation preconditioning. In pancreatic adenocarcinoma models, low-dose radiation therapy was effective at sensitizing tumor cells to locally activated CAR T-cells. Low-dose radiation prevented antigen escape and induced expression of chemokine ligand, which is a cell-derived factor which stimulates T-cell migration and adhesion to the activated endothelium.63,64 Together, these mechanisms effectively sensitized these antigen-negative cells to TRAIL-mediated death. 65 Thus, low-dose radiation therapy may provide effective sensitization as part of a CAR conditioning regimen. Given the ‘synergistic’ effects chemotherapy and T-cell redirecting immunotherapy have on the immune contexture, it would be reasonable to explore strategies in which cytotoxic chemotherapy could be an adjunctive treatment with BiTE therapies and could transcend a role merely as conditioning therapy for CAR T-cells.
Further investigation into the optimal positioning of T-cell redirection therapy within the metastatic PCa treatment cascade is warranted. The immune contexture within the TME evolves with disease progression, and it is possible that earlier utilization of CAR T-cell and BiTE therapies will allow for more robust and durable immunologic responses. Advanced disease harbors a chronically inflamed TME with a cellular predominance of TAMs and MDSCs as well as cytokines which promote tumor cell proliferation, epithelial-to-mesenchymal transition, and resistance to traditional chemotherapeutics. 66 Altogether, this microenvironment is a barrier to efficacy of both traditional cytotoxic chemotherapy and also novel T-cell immunotherapies. Cytotoxic chemotherapy as a class is further limited by cumulative systemic toxicities and has less potential for a sustained response. Therefore, patient selection will be paramount to revealing the true potential of these agents and there is biologic rationale to transitioning away from a heavily pretreated patient population in future clinical study.
Finally, the question remains as to whether to limit PSMA-targeted T-cell therapy to patients with evidence of radiographic PSMA avid disease, as is the case in theranostics with 177Lu-PSMA-617. Even if there is radiographic improvement with treatment, durability of response will rely on T-cell trafficking into the TME, persistence, and effector T-cell function. While there is certainly credence to radiographic-based precision medicine, tailoring therapy by molecular phenotype will likely yield more optimal outcomes with T-cell immunotherapies. 67 Molecular phenotyping of tumors is not limited to assessment of TAAs, but rather can include immunophenotyping of tumor-infiltrating immune cells which can inform optimal timing and treatment choice of T-cell therapies.
Perspectives on drug design
CAR engineering strategies continue to evolve in response to new challenges with CAR implementation in solid tumor oncology. Modifications to the number and type of stimulatory domains, armoring domains, and optimal utilization of viral vectors or nonviral transposon-based systems remain under study. Recently, natural killer (NK) cells have shifted attention away from T-lymphocytes as CAR drivers with proven tumor-targeted activity and limited risk of toxicity. CAR-engineered NK cells pose major theoretical advantages over conventional T-cells. Irrespective of CAR-expression, NK cells have the natural ability to exert cytolytic effects on target cells through various TAA-unrestricted receptors as well as through FcγRIII antibody-dependent cell-mediated cytotoxicity. CAR transduction in allogeneic NK cells provides the theoretical ability to express dual-target antigen-expressing domains in addition to their natural spontaneous cytotoxic functions. NK cells provide further advantage over T-lymphocytes because of their limited life span, thereby potentially lessening the risk of sustained toxicity and CRS, given these cells are rapidly cleared from circulation. Anti-PSMA CAR-engineered NK-92 cells evaluated in vitro and in PCa xenograft models demonstrated antigen-specific functionality, cytolytic activity including effective interferon-γ (IFNγ) production.3,68
Whereas the prolonged survival of PSMA CAR T-cells provides issues with toxicity, the short serum half-life of prototypical BiTEs contributes to immunogenicity and toxicity. 13 Efforts to extend BiTE half-life in circulation have largely centered on inclusion of additional Fc domains to the bsAb format. This strategy has successfully extended drug half-life in circulation as evident by our experience with AMG160, the first HLE PSMA-targeting BiTE. However, this strategy comes at risk of increased toxicity. Inclusion of additional Fc domains risks inadvertent cross-linking of standard Fc fragments and aggregation with adjacent antibody leading to off-target T-cell activation and CRS. 69 It is possible that the ‘small-molecule’ BiTE consisting of two covalently linked single-chain fragments is a suboptimal antibody format. Emerging data support the use of larger IgG-based bispecific molecules (IgGsc) as an alternative T-cell redirection modality to streamline drug delivery. In PSMA-expressing PCa models, serum half-life of the IgGsc molecule was much longer with a considerable amount of antibody detected intratumorally 48 h after injection. Impressive antitumor efficacy was noted in vivo with minimal unwanted off-target T-cell activation and cytokine release. 70 Ultimately, development of bsAb formats with pharmacokinetic properties which maintain sufficient serum levels of drug, such as PEGylated or albumin-bound molecules, is likely to overcome barriers observed in trials with prototypical BiTE constructs. 71
The impressive preclinical and clinical findings of dominant-negative TGF-β CAR T-cells serve as a proof-of-principle that ‘armoring’ motifs confer superior T-cell responses. Narayan et al. 46 provide mechanistic insight into how the armoring domain directly impedes immunosuppressive TGF-β signaling. This study underscores the need for further development of armoring moieties in future generation CAR design. TMunity has now engineered a double-armored CAR with a CD2 endodomain and a novel PD1-CD28 switch fusion protein as an additional armor to overcome PD-L1-induced T-cell anergy (PSMA-CD2z:dnTGFBR:PD1-CD28). Preliminary studies demonstrated feasibility of PSMA-CD2z:dnTGFBR:PD1-CD28 production, cytotoxicity, memory profile, and activation while having a significantly reduced risk of off-target immune activation. 72 Investigators are also now engineering fourth-generation triple-armored constructs harboring cytokine-secreting moieties which modulate the immune milieu and in turn foster an immunologically ‘active’ resident immune cell population to augment CAR T-cell function. 73 TmPSMA-03 is a PSMA-directed fourth-generation CAR now in preclinical development. 74 Based on this ongoing success of dominant-negative signaling in CAR development, investigators have devised an innovative platform which exploits tumor-derived TGF-β signals into immune-stimulatory signals. The CAR chimeric TGF-β receptor (CAR-CTBR) T-cells express chimeric variants of the TGF-β receptor in which the receptor is fused to the transmembrane and intracellular domains of immunostimulatory IL-12 receptors (IL-12R-β2 and IL-12R-β1). Exposure of CAR-CTBR T-cells to TGF-β had a stimulatory effect on cell expansion, cytokine production, and T-cell effector function with upregulation of IFNγ, IL10, IL18RAP, IL18R1, IL21R, and CD62L transcripts. 35
Perspectives on drug toxicity
Reducing immune toxicity is necessary for delivery of therapeutically active doses of drug. Our understanding of CAR T-cell and BiTE therapy toxicity is informed by the hematologic malignancy experience with these classes of agents. Toxicities have conventionally been believed to be unpredictable and potentially severe. However, the observed toxicity of PSMA CAR-T and BiTE therapy in PCa thus far has been predictable. Any-grade CRS is nearly ubiquitous, and aside from the experience from select CAR T-cell therapies, toxicities have been managed effectively. The PSMA × CD3 BiTEs have not demonstrated any significant target-related adverse events, and PSMA expression on nonprostatic tissue has not been a barrier to drug delivery. There were fatalities observed in three recent phase I PSMA-targeted CAR T-cell therapy trials, although not all deaths were definitively treatment related. Correlative analyses in these patients confirmed a similar pathophysiologic mechanism underlying CRS in PCa as in hematologic malignancies, with an observed elevated inflammatory signature with higher levels of IL2, IL6, GM-CSF, and IL-18. As such, we advocate for early pharmacologic intervention at the first sign of CRS, including pyrexia, as is established within the hematology literature. As to whether high-dose corticosteroids or tocilizumab (IL6-R antagonist) blunts the elicited T-cell response remains unclear in PCa, but emerging data suggest there is no effect on treatment efficacy or drug kinetics and that early intervention to attenuate toxicity improves outcomes.75–77 Study protocols are now incorporating prophylactic doses of tocilizumab as a preventive measure and it is likely that with these and other mitigation measures, we may achieve greater dose thresholds and achieve improved clinical responses.78,79
Perspectives on modulating the TME
The advent of T-cell redirection as a therapeutic class introduces levels of complexity to drug discovery that have yet to be reconciled. As we gain a deeper understanding of the effects our therapies have, and barriers they face within the TME, we continue to adapt our drug engineering to include alternative domains, signaling motifs, and structural changes for optimal design therapeutic windows and adequate antitumor activity. However, even an optimized CAR or BiTE may be insufficient to account for the multitude of variables and hindrances presented by the complex and immunosuppressive TME. 27 The PCa TME is unique in degree of hypoxia, low pH, high extracellular potassium levels, and tumor stroma rich in fibroblasts, resident T-regulatory cells, M2 TAMs, and MDSCs, all of which lead to insufficient influx of effector T-cells which in turn leads to challenges achieving sufficient dosing for antitumor activity. 80 Multimodal therapy incorporating cellular immunotherapy with other therapeutic avenues such as ICB, cytotoxic chemotherapy, antiandrogen therapy, or radiotherapy may overcome these barriers and effectively convert the ‘cold’ PCa immunophenotype into a ‘hot’ immunogenic tumor. 81
Combination strategies may be paired with biomarker-based assays to assist in patient selection and for tailored therapy. Integration of spatial immunoprofiling assays which characterize T-cell populations from freshly allocated tissue for patients being considered for T-cell immunotherapy may surmount barriers of immune escape. Depending on the T-cell phenotype, whether exhausted or senescent, targeted approaches can be employed to convert immunologically inactive resident cells into ‘sensitized’ cells. 82 Adjunctive treatments under investigation include indoleamine 2,3-dioxygenase (IDO) inhibitors, antibody agonists to costimulatory ligands such as CD28 and CD137, ICB, and soluble cytokines. 69 Multiple studies combining ICB with BiTE therapy are well underway, and we now have an abundance of preclinical data which provide rationale for ICB–CAR T-cell combination strategies. Soluble immune-activating cytokines also have the potential to alter the TME and lower the threshold for effective T-cell infiltration and activation. Cytokines have pleotropic abilities which affect the stroma barrier, compromise the extracellular matrix and surrounding fibroblasts, create a permeable basement membrane for T-cell trafficking, and promote expansion of peripheral T-cells. CAR T-cells engineered to inducibly express and release IL-18 were effective at fostering an inflamed TME with reduction in Treg and M2-polarized cells in immunocompetent solid tumor mouse models. 83 In evaluation of early-generation PSMA-targeted CAR T-cells, Junghans et al. 43 implemented adjunctive low-dose IL-2 in the study design. Treatment efficacy positively correlated with IL-2 levels. Indeed, in patients with suboptimal treatment effect, there is a perception that CAR T-cell activity was hindered by low plasma IL-2 levels having been depleted by high levels of engrafted T-cells, ultimately being a limiting factor to achieving the best CAR T-cell antitumor activity. Adjunctive immune-activating cytokine strategies may benefit future trial design for both CAR T-cells and BiTEs for patients with PCa.
Alterations to the TME are not limited to pharmacologic interventions with adjunctive therapies. Genomic editing techniques may soon have a role in accelerating T-cell immunotherapy efforts. CRISPR/Cas9 technology has been integrated into CAR genome editing as a means of disrupting microenvironment inhibitory signals. Gene editing can effectively knock out inhibitory ligands or introduce dominant-negative domains to create CAR T-cells resistant to inhibitory pathways. Oncolytic adenoviruses may soon be employed as cancer virotherapy provides an alternative method to disrupt TME inhibitor signals. Oncolytic adenoviruses selectively infect and replicate in malignant cells and can be genetically engineered to express transgenes which may have synergistic potential with T-cell immunotherapy. 84 Prior studies demonstrated the feasibility of utilizing oncolytic adenoviruses as a vehicle for delivery of immune mediators which increased T-cell trafficking and prolonged effector T-cell survival on tumor-bearing mice. 85 Investigators have developed a novel platform to engineer genetically tailored oncolytic adenoviruses which secrete ‘endogenous’ BiTEs upon viral replication in cancer cells. 86 The BiTE-expressing oncolytic adenoviruses demonstrated robust T-cell activation and proliferation in mouse xenograft models. These data provided rationale for combination therapy of BiTE-expressing oncolytic adenoviruses with CAR T-cell therapy as a means to overcome the immunosuppressive barriers and immune escape intrinsic to CAR-T and BiTE monotherapies. In a recent preclinical report, investigators combined CAR T-cells targeting folate receptor alpha (FR-α) with oncolytic adenoviruses armed with epidermal growth factor receptor (EGFR)-targeting BiTEs in xenograft tumor. The combination of virus, BiTE, and CAR T-cells leads to synergistic potentiation of the immune response with more pronounced and durable antitumor activity compared with each monotherapy. The enhanced antitumor activity of BiTE and CAR T-cells is likely synergistic as CAR T-cells can be engaged through BiTE-CD3-mediated activation as well as CAR T-cell antigen target recognition. Wing et al. 87 elegantly demonstrated that this model can exploit the effector potential of CAR-negative cells found in CAR T-cell preparation. BiTE-expressing oncolytic viruses can redirect CAR T-cells toward secondary antigen in the absence of expression of CAR-targeted antigen. BITE-armored adenovirus constructs are under rapid development in various tumor models, and future combination therapy strategies employing PSMA CAR T-cells and BiTE constructs may be advantageous and warrant exploration.
Conclusion
T-cell redirecting therapies have a relatively short natural history, but as a therapeutic class, they are poised to have transformative impact on the treatment of PCa. Emergence of PSMA as a targeted ligand and the recent success of PSMA in theranostics are hypothesis generating and provide a framework for ongoing study in cellular therapy. This enthusiasm is now leading to drug development with attention to other TAAs highly specific to PCa, including PSCA and STEAP1. T-cell immunotherapies targeting multiple TAAs can lead to improved immunogenicity and outcomes. Drug delivery has conventionally been resource intensive, limiting study activation thereby limiting recruitment. However, toxicity management recommendations are now more streamlined with prior experiences informing effective safety measures within protocols. Altogether, we are now witnessing rapid development of novel iterations of T-cell immunotherapies and early-phase trials, with newer generation constructs having greater immunogenicity and improved therapeutic windows. The field of T-cell immunotherapy in PCa will likely continue to prosper and there is promise for breakthrough in the near future.
The preclinical data surrounding PSMA-targeted CAR T-cells and BiTEs are encouraging, and the negative clinical trials to date do not constitute failure. In the studies reported thus far, responses vary but meaningful antitumor activity is clear and consistent. In rare instances, responses are compelling. Clarifying the molecular underpinnings of the rare responses through correlative analyses from ongoing and future studies will inform more efficacious therapeutic strategies, likely in the form of combination therapies. PMSA-directed CAR T-cell therapy and BiTEs face similar barriers to successful implementation, including a hostile and highly suppressive tumor immune contexture which prevents T-cell-mediated cytolysis and difficulty achieving an effective dosing regimen without undue toxicity. As we learn from clinical experiences of older generation CAR T-cell and BiTE constructs, and pairing these experiences with our increasingly nuanced understanding of barriers, we are poised to develop rational CAR and BiTE design tailored to the complexities of the PCa TME.