
Abstract
Chronic subdural hematoma (CSDH) is an increasingly common disorder characterized by persistent accumulation of blood products and inflammatory exudate within the dural border cell (DBC) layer. Its pathogenesis represents a self-sustaining cycle of inflammation, pathological angiogenesis, and impaired resolution. Injury of the head, which may be traumatic or nontraumatic, initiates cleavage of the DBC layer, leading to fibroproliferative membrane formation mediated by collagen synthesis and TGF-β1/SMAD signaling. The resulting outer membrane develops fragile neovasculature, driven primarily by vascular endothelial growth factor, facilitating recurrent microhemorrhage and fibrinolysis perpetuating hematoma expansion. Chronic inflammation sustains disease progression through macrophage polarization, cytokine release, and increased vascular permeability, processes amplified by age-related immune dysregulation and hypoxia-induced factor-1alpha signaling. Impaired tissue repair due to metabolic deficits further limits resolution. Concurrent dysfunction of meningeal lymphatic drainage and fibrotic arachnoid granulations compromises clearance of blood degradation products and inflammatory mediators, while matrix remodeling and cerebrospinal fluid ingress contribute to hematoma persistence. This narrative review presents a pathophysiologic framework highlighting CSDH as a dynamic inflammatory-angiogenic disorder. Pharmacological strategies targeting inflammation, angiogenesis, fibrinolysis, hypoxia, and matrix remodeling hold potential as complements or alternatives to established treatments, including surgical drainage and middle meningeal artery embolization. As the burden of CSDH on healthcare systems rises, translational research and controlled clinical trials will be critical to developing mechanism-driven, multimodal management paradigms.
Plain language summary
This manuscript fits squarely within the scope of Therapeutic Advances in Neurological Disorders as it provides a comprehensive, mechanistic review of chronic subdural hematoma—a condition projected to become the most common cranial neurosurgical disorder by 2030. By integrating anatomical, molecular, and clinical perspectives, it highlights the pathophysiological processes that underpin disease chronicity and recurrence, while also linking these insights to evolving surgical and adjunctive treatment strategies. The work is directly relevant to neurosurgeons, residents, and students as it addresses both operative considerations and future directions in therapeutic innovation, thereby contributing to the advancement of evidence-based neurosurgical care.
Keywords
Introduction
Background and clinical context
Chronic subdural hematoma (CSDH) is characterized by the persistent accumulation of blood products and inflammatory fluid within the dural border cell (DBC) layer of the dura mater. 1 First described by Virchow in 1857, 2 it predominantly affects older adults and is associated with a pathological cycle of inflammation, recurrent microhemorrhages, and membrane formation. 3 Over the past few decades, the incidence of CSDH has increased significantly, with rates ranging from 1.7 to 20.6 per 100,000 individuals annually. 4 Projections indicate that CSDH will become the most common cranial neurosurgical condition among adults by 2030, underscoring its rising impact on healthcare systems worldwide. 5
Clinically, CSDH presents a diagnostic challenge due to its nonspecific symptoms, which typically develop insidiously over weeks to months. Patients may experience headaches, cognitive decline, or focal neurological deficits. Diagnostic imaging is crucial, with computed tomography (CT) being the primary modality, supplemented by magnetic resonance imaging (MRI) when necessary.4,6 Symptom onset is often preceded by a minor head injury that disrupts the DBC layer—an event so subtle it may go unnoticed by the patient. 7 Nontraumatic factors associated with CSDH include cerebral atrophy of advanced age, warfarin and aspirin therapy, cerebral amyloid angiopathy, subdural hygroma, and intracranial hypotension from spinal cerebrospinal fluid (CSF) leakage.8–12
Historically, CSDH was attributed to the rupture of bridging veins following minor trauma. 7 However, emerging evidence now redefines CSDH as a chronic inflammatory condition driven by angiogenesis and persistent membrane activity. This narrative review synthesizes the latest advances and insights in the pathophysiology of CSDH, outlining the sequence of pathological events that drive its progression. By elucidating the molecular and cellular mediators involved, this comprehensive overview highlights potential therapeutic targets that may guide the development of new treatment strategies in response to the anticipated increase in disease burden.
Anatomical considerations
The meninges are the protective covering surrounding the brain and spinal cord, consisting of three primary layers: the dura mater, the arachnoid mater, and the pia mater (Figure 1). The dura mater, the outermost meningeal layer, is itself composed of several sublayers that each play distinct roles in cerebrovascular anatomy and injury response. The outer periosteal layer adheres tightly to the inner surface of the skull, while the inner meningeal layer lies adjacent to the arachnoid membrane. 13 Between them is the DBC layer, consisting of flattened fibroblast-like cells and large extracellular spaces filled with an amorphous substance. The DBC layer is relatively structurally weak and susceptible to cleavage or delamination, serving as the site for the formation of CSDH.1,14

Diagrammatic representation of the anatomy of the meninges. The dural border cell layer—structurally weak and collagen-poor—is the site of pathology.
Importantly, the dura mater houses a network of meningeal lymphatic vessels (MLVs), which facilitate immune surveillance and drainage of CSF into the deep cervical lymph nodes.15,16 Impairment of MLV function has been implicated in many central nervous system diseases, including CSDH. 17
In contrast to blood vessels in the brain parenchyma, meningeal blood vessels lack an ensheathing glial barrier or glia limitans. This functional absence makes them more vulnerable to vascular leakage. 18 In CSDH, this structural difference promotes persistent inflammation and impaired resolution. The MMA serves as the primary vascular supply to the dura. Given the MMA’s dominant role, it is the target for therapeutic embolization to disrupt the cycle of pathological angiogenesis. 19
Beneath the dura lies the arachnoid. Its outer arachnoid barrier layer, sealed by tight junctions and a basement lamina, forms the true physiologic boundary of the CSF compartment. The arachnoid barrier layer abuts the weaker DBC layer, a collagen-free zone that lacks tight junctions and contains irregular extracellular cisterns. 14 The outermost arachnoid and the innermost dura are tightly attached to each other. In the normal state, no subdural space exists. Rather, trauma or pathology cleaves within the DBC layer due to its lack of coherence, creating the plane into which blood dissects, and chronic subdural collections organize. Thus, while the arachnoid barrier layer maintains integrity during traction, the structurally fragile DBC layer provides the tissue plane that allows CSDH to develop.
Sequence of pathological events
Initiating event: Injury/trauma
Following minor trauma, CSDH typically takes 4–7 weeks to develop noticeable symptoms. 20 Although most patients with mild traumatic brain injury (TBI) recover from meningeal vascular damage within 3 weeks, a subset fails to resolve. 21 Gross examination often reveals encapsulated collections of blood and proteinaceous fluid within the DBC layer. These hematomas frequently exhibit internal septations, a sign of repeated hemorrhages and resorption processes.22,23 MRI commonly demonstrates heterogeneous signal intensities on T1- and T2-weighted images due to blood at different stages of degradation. Triangular-shaped contrast enhancement at the hematoma margins is a characteristic finding.24,25 Contrast enhancement is also consistently observed along the outer membrane, reflecting ongoing inflammation.
Separation of DBCs and membrane formation
The inciting injury leads to a tear in the DBC layer, which may not immediately cause bleeding but creates a potential space that, over time, fills with blood and fluid due to inflammation, neovascularization, and delayed microhemorrhages (Figure 2). In the microscopic findings of CSDH, the periosteal and meningeal layers of the dura mater remain histologically unchanged, while injury is observed within the DBC layer. In control individuals, the DBC layer is thin and contains minimal fibrous tissue. However, after injury, this layer thickens due to the increased presence of collagen fibers and fibroblast proliferation.26,27

This diagram illustrates the anatomical layers of the meninges and the time-dependent pathophysiological events leading to CSDH. Minor trauma induces injury to the dural border cell layer, initiating inflammation and neovascularization within days. Over the course of a week, fibroblast activity, vascular remodeling (via VEGF, PDGF, uPAR), and cytokine release (IL-6, IL-8) result in membrane formation and persistent subdural fluid accumulation.
CSDH fluid contains high levels of type 1 (PICP) and type 3 (PIIINP) procollagen, suggesting a fibro-proliferative process similar to wound healing. 27 The separation of DBCs initiates this repair, but excessive collagen synthesis results in pathological membrane formation. This process is mediated by the SMAD signaling pathway, activated by TGF-β1 from eosinophils. 28 Membranes form along the arachnoid and dura mater, known as the inner and outer membranes, respectively.
Inner membrane: The inner membrane is composed of four distinct layers. First is the hematoma surface, followed by the intermediate layer, which occasionally contains eosinophils and edematous fluid in the expanded extracellular space. Next is the arachnoid surface layer, where blood pigments, fibrin, and fibrinoid material are interspersed with loosely arranged collagen fibers and elastin. Finally, the innermost layer consists of cells that exhibit few of the tight intercellular junctions, such as desmosomes, typically found in the arachnoid mater. 29 The inner membrane is not the major contributor to hematoma expansion.
Outer membrane: The outer membrane contains numerous capillaries with wide lumens. The walls of these capillaries are composed of a single layer of flattened endothelial cells with a faint or poorly defined basement membrane. Notably, the vascular walls generally lack smooth muscle cells with an incomplete surrounding layer of pericytes. These capillaries exhibit increased permeability to red blood cells, fibrin, and plasma due to large gaps between cells, allowing vascular contents to easily leak into the surrounding extravascular space. 30
These membranes represent a transformation of the DBC layer into a chronically inflamed tissue. Therefore, repetitive subdural hemorrhages are the consequence rather than the cause of membranes.
Inflammation: Sustained immune response
In 1925, Putnam and Cushing postulated that Kupffer cells—also known as stellate macrophages—were involved in the phagocytosis of erythrocytes in CSDH, based on the green, bile-like discoloration observed in subdural clots. 3 Nearly a century later, advances in single-cell sequencing technologies have substantiated and expanded upon this early hypothesis, offering a detailed view of the immune landscape within CSDH lesions. Immune profiling reveals elevated classical dendritic cells (cDC2) and M2 macrophages in CSDH lesions, with high reactive oxygen species (ROS) expression and IL-1β signaling promoting anti-inflammatory macrophage recruitment. 31
Because CSDH often arises as a delayed complication of minor TBI, insights into TBI-related immune responses provide a valuable framework for understanding CSDH pathophysiology. Following TBI, there is a marked increase in “Activated Macrophages 1,” characterized by the high expression of complement-related genes, as well as a significant rise in fibroblast populations. Macrophage ligands like Tgfb1 stimulate cell growth and differentiation. Other macrophage ligands (Itgam, Apoe, Vcam1, Selplg, Nectin1, and Itgb1) facilitate cell adhesion and phagocytosis. 26 Concurrently, fibroblasts upregulate immune system activation and cytokine signaling genes, with markers like Foxc2 and Fxyd5 significantly elevated post-injury. Increased fibroblast activity also correlates with enhanced collagen density in the meninges. Additional fibroblast ligands (Apoe, Csf1, Cxcl12, and Tgfb3) support tissue growth and inflammation. 26
Shortly after TBI, vascular damage and meningeal cell death result in the generation of ROS, compromising the glial limitans and propagating neuronal death into the adjacent brain parenchyma. In experimental models, transcranial administration of glutathione, an ROS scavenger, within 3 h of injury reduces parenchymal cell death by up to 67%, preserves the glial limitans, and prevents further damage. 32 These protective effects suggest that glutathione and similar antioxidant strategies may have therapeutic potential in the treatment of CSDH.
In murine models of mild TBI, single-cell RNA sequencing shows increased expression of Type I interferon signature genes (Ifnar1, Ifi203, Irf2bp2, and Irf5) in macrophages. Interestingly, bulk RNA sequencing reveals that, while young mice return to baseline gene expression 1.5 months post-injury, aged mice continue to exhibit elevated levels of Type I interferon-related genes (Ifit1, Irf7, and Mx1), contributing to chronic inflammation in older meningeal environments. 26 This impaired immune resolution in aged meninges may help explain the higher CSDH incidence in elderly populations.
The immune response involves temporal shifts: acute inflammatory macrophages give way to wound-healing macrophages expressing anti-inflammatory ligands. The immune response begins when compression injuries induce meningeal macrophage death and the formation of a “honeycomb” network of microglia at the glial limitans. These microglia undergo transformation into motile, phagocytic “jellyfish” shapes. 32 Inflammatory phase macrophages express adhesion molecules and chemotaxis-related genes, including Ccr7, Ccl22, and Ccl5.33,34 The acute inflammatory phase is characterized by beneficial immune responses—such as neutrophil swarming and microglial repopulation—but secondary insults during this phase may exacerbate injury and impede tissue repair.21,32 One week after injury, meningeal macrophages exhibit a shift toward wound healing and inflammation resolution, with increased frequencies of “Anti-Inflammatory” and “Resolution Phase” healing macrophages. 26 Therefore, both phases of the immune response are needed for repair.
The multiphase and mixed process of wound healing is evident in the activity of T cells. T cells in the inflammatory environment upregulate genes associated with survival (Gimap1 and Gimap6), activation (Arrb2, Ppia, and Cd48), cytokine signaling (Mif and Il2rg), and antigen recognition (Cd247). At the same time, these cells express immune-dampening genes such as Socs1 and Cd52, reflecting a nuanced immune response. 26 Anti-inflammatory pathways are evident, with ligands found like Anxa1, which inhibits adhesion and migration, and Serping1, which induces C1 inhibitor production. 26 Patients with recurrent CSDH exhibit a higher white blood cell count in subdural fluid during the second surgical procedure compared to the first. 35 This suggests persistent or heightened inflammatory activity associated with recurrence risk.
Osteopontin (OPN), a glycoprotein induced by injury in various tissues, plays a significant role in the promotion of CSDH. OPN is an extracellular matrix protein that undergoes thrombin-mediated cleavage, producing an N-terminal fragment that plays a key role in activating integrin-mediated signaling pathways. The concentration of the N-terminal half of OPN is significantly higher in CSDH fluid than in serum. The N-terminal half of OPN activates integrin α9 and β1 signaling pathways, promoting leukocyte recruitment and fibroblast activation.36,37 In addition, OPN and vascular endothelial growth factor (VEGF) co-expression in the CSDH membrane further amplifies integrin signaling and vessel remodeling. 36 OPN’s role extends to microglial phagocytosis, highlighting its relevance in CSDH’s chronic inflammatory environment. 38
Neutrophil extracellular traps (NETs) have also been implicated in the inflammatory processes associated with CSDH, contributing to its chronic nature. 39 NETs are networks of extracellular fibers composed of DNA, histones, and proteases expelled by neutrophils during infections and inflammatory responses. Table 1 summarizes the key agents in the promotion of inflammation.
Summary of key players in the promotion of CSDH inflammation.

CSDH, chronic subdural hematoma; OPN, osteopontin.
Angiogenesis: Formation of fragile blood vessels
The neovascularization in CSDH is tightly linked to the inflammatory and repair responses triggered by meningeal injury. 29 Matrix metalloproteinase-2 (MMP-2), secreted by fibroblasts, is an enzyme that degrades extracellular matrix components, allowing remodeling of vascular walls and surrounding connective tissue. 40 The extracellular matrix acts as a storage compartment for angiogenic growth factors such as VEGF, which are released from sequestration by proteolytic degradation of the extracellular matrix. 41 Fibroblast ligands such as CXCL12 and placental growth factor (PGF) also promote angiogenesis. CXCL2, a chemokine primarily involved in neutrophil recruitment, and PGF cooperate with vascular growth factors such as Angiopoietin-1 (Ang-1) and Angiopoietin-2 (Ang-2) to modulate the stability and permeability of newly formed blood vessels. 42
Neovascularization relies on both extravascular fibrin clearance and fibrin deposition as a provisional matrix component to support cell migration and tissue remodeling.1,14,43 Endothelial cells secrete platelet-derived growth factor (PDGF), which attracts pericytes, inducing their attachment to new blood vessels.44,45 Neovessel endothelial cells themselves also express VEGF. 45 Co-expression of VEGF and PDGF receptor (PDGF-BB) induces angiogenesis. 46 Concentrations of PDGF-BB in CSDH fluid are 41 times higher compared to CSF. 47 Therefore, blocking PDGF-BB presents therapeutic potential for reducing angiogenesis in CSDH. Table 2 summarizes the key agents in the promotion of angiogenesis.
Summary of key players in the promotion of CSDH angiogenesis.
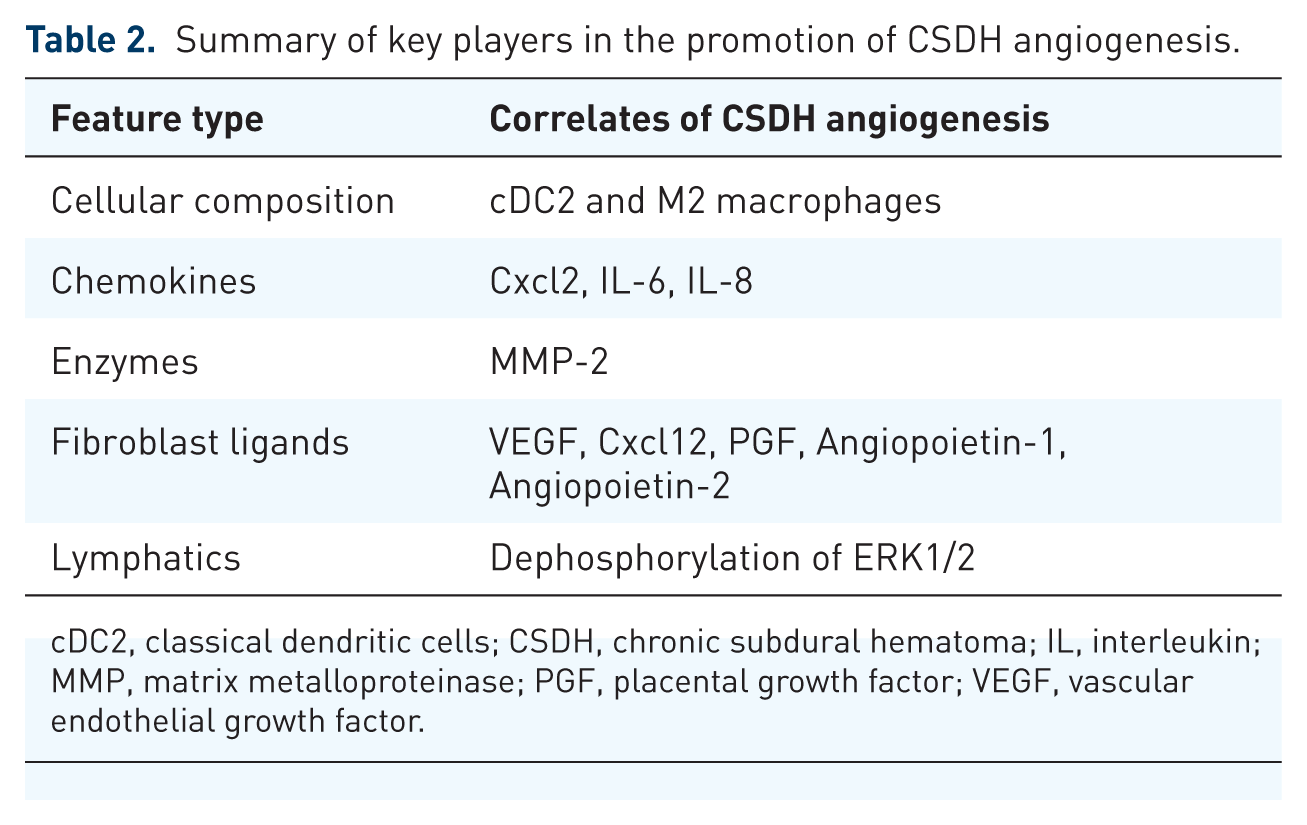
cDC2, classical dendritic cells; CSDH, chronic subdural hematoma; IL, interleukin; MMP, matrix metalloproteinase; PGF, placental growth factor; VEGF, vascular endothelial growth factor.
VEGF plays a central role in neovascularization by promoting the growth of fragile, leaky blood vessels in the outer membrane, which leads to repeated microhemorrhages and fluid exudation.36,48 The inflammatory milieu in the hematoma—driven by blood degradation products (e.g., thrombin, fibrinogen degradation products), hypoxia-inducible factor (HIF), cytokines like interleukin-6 (IL-6) and tumor necrosis factor-alpha, and cyclooxygenase-2 activation—upregulates VEGF expression.49–56 This creates a pro-angiogenic environment, where VEGF is secreted locally to induce new vessel formation.
VEGF is found in extremely high concentrations in the hematoma fluid (often dozens of times higher than in serum). 57 mRNA expression of VEGF isoforms is elevated, particularly in cells from the hematoma fluid and inconsistently in membrane tissue, suggesting dynamic secretion.42,58 Secreted VEGF binds to receptors on endothelial cells, activating pathways like PI3-kinase/Akt and Ras/MEK/ERK, which promote endothelial proliferation.36,56 The imbalance with other factors like Ang-2 (overexpressed in membranes) over Ang-1 further destabilizes vessels, amplifying VEGF’s effects.42,59–62 Therapeutic interventions such as corticosteroids or statins target this by inhibiting VEGF expression and reducing membrane-driven angiogenesis.63,64
VEGF production in CSDH membranes is driven by a combination of resident and infiltrating cells responding to the local inflammatory and hypoxic environment. Macrophages are the primary VEGF producers, activated by hematoma breakdown and cytokines, and correlate with PGE2 and VEGF levels.48,58 Endothelial cells amplify angiogenesis through VEGF signaling and fragile microcapillary formation, while plasma cells sustain VEGF via immune responses. 65 Mesenchymal cells help form the outer membranes and extracellular matrix, and neutrophils/mononuclear cells dominate early hematoma fluid, fueling initial VEGF release. 42 Together, these cells maintain chronic inflammation, vessel growth, and membrane persistence. The outer membrane is rich in these cells, which express high levels of VEGF mRNA and protein. 66 Key producers of VEGF are shown in Table 3. This process highlights CSDH as an “angiogenic disease,” where membrane-derived VEGF drives pathology. Clinically, anti-VEGF agents such as bevacizumab are under investigation and show promise as a potential treatment for CSDH in select patients.67–70
The major cell populations present in CSDH membranes and fluid, their stimuli for activation, and their contributions to VEGF production through distinct but overlapping mechanisms.

CSDH, chronic subdural hematoma; HIF, hypoxia-inducible factor; IL-6, interleukin; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor.
Fibrinolysis and recurrent bleeding
In CSDH, the local hemostatic balance is shifted toward persistent fibrinolysis within the outer membrane, compromising clot stability and perpetuating the lesion. Tissue plasminogen activator (t-PA) released from endothelial cells increases local fibrinolysis, disrupting clot stability and impairing the clotting cascade. 39 This process contributes to recurrent bleeding and partial vessel occlusion within the outer membrane of the hematoma. 71 Blood vessel distribution varies between cases and fields, supporting the theory that vascularization is an active response to injury. 1
Urokinase plasminogen activator receptor (uPAR) plays a significant role in fibrinolytic processes. uPAR binds to urokinase plasminogen activator, facilitating the conversion of plasminogen to plasmin, which breaks down fibrin clots and activates MMPs that lyse connective tissue within the hematoma. 72 Hematoma fluid from patients with CSDH contains elevated median uPAR levels (22,125 pg/L), far higher than systemic blood levels (789 pg/L). 73 This pathophysiological framework supports the rationale for adjunctive therapies targeting the fibrinolytic axis.
Fluid accumulation and hematoma expansion
Following TBI, damage to the glial limitans may permit excessive fibrin infiltration into the brain parenchyma, exacerbating local inflammation. In TBI models with monocyte depletion, persistent fibrin deposition has been observed, accompanied by impaired revascularization. 21 This pathological process can lead to basement membrane thickening and local hypoxia, potentially perpetuating a cycle of inflammation and vascular dysfunction. This underscores how early treatment after TBI can have lasting structural consequences.
Recent evidence also suggests that CSF plays a significant role in hematoma expansion. For example, in a study analyzing hematoma contents, β-trace protein (βTP)—a highly specific marker for CSF—was found in 93% of subdural fluid samples, indicating frequent admixture of CSF within the hematoma cavity. 74 This finding implies that arachnoid breaches or dural permeability may allow CSF to infiltrate the subdural space, contributing to the osmotic gradient that drives further hematoma expansion. The presence of CSF is particularly prominent in recurrent CSDH cases, suggesting its role in promoting chronicity. This points to a mechanical–biochemical mechanism of recurrence in CSDH. Beyond leaky vessels, CSF entry into the hematoma cavity may serve as a persistent volumetric driver, undermining the effects of surgical evacuation.
Hypoxia: Oxygen deprivation and metabolic changes
The mass effect of the CSDH is suggested to reduce regional cerebral blood flow, producing perfusion deficits more pronounced in the central cerebral areas, leading to reduced oxygen delivery.72–74 Local brain perfusion autoregulation remains active in the cortical area underlying CSDH, with significant upregulation of cerebral blood volume and, consequently, cerebral blood flow reflecting compensation in tissue at risk of ischemia. 75 Despite this autoregulatory response, oxygen deprivation may stimulate an increased vascular supply to the affected area. VEGF and Hypoxia-Inducible Factor-1alpha (HIF-1α) play central roles in vascular remodeling. Under hypoxic conditions, VEGF expression is upregulated, promoting the formation of new blood vessels.49,65,66,76,77 The middle meningeal arteries of the periosteal layer and penetrating arteries of the meningeal layer supply the proliferation of arterioles and leaky capillaries to the DBC layer.19,30 Imaging studies reveal that arterioles in this region proliferate more than fivefold.
Hypoxia may also be exacerbated by increased central venous pressure, which disrupts claudin-5—a critical protein maintaining tight junctions between neural endothelial cells. 78 This disruption weakens capillary integrity, making them more prone to leakage and bleeding. In addition, IL-6 can widen endothelial gap junctions via activation of the JAK/STAT3 pathway, further increasing vascular permeability.79,80
Chronic hypoxia may stimulate a metabolic shift toward increased fatty acid oxidation, an alternative energy pathway in which fatty acids are broken down to meet cellular energy demands. 81 During this process, acylcarnitine is generated to facilitate the transport of fatty acids into the mitochondria, where they undergo oxidation to produce ATP. 82 However, in patients with recurrent CSDH, immunometabolic analysis of the hematoma fluid reveals a 35% decrease in acylcarnitine levels, along with deficiencies in acyl-CoA dehydrogenases—key enzymes involved in fatty acid oxidation. 83 These findings suggest that impaired fatty acid oxidation may contribute to the recurrence of CSDH. Inadequate fatty acid oxidation could limit the energy available for reparative processes within the hematoma membrane, perpetuating a pro-inflammatory, hypoxic microenvironment. This metabolic insufficiency may weaken local vascular stability and impair collagen remodeling—suggesting a potential therapeutic target in energy metabolism modulation.
Impaired lymphatic drainage
In CSDH, lymphangiogenesis increases over 50-fold in the periosteal and meningeal layers. 84 However, despite this proliferative response, meningeal lymphatic drainage dysfunction is strongly associated with higher CSDH recurrence rates.85–87 Basal MLV dysfunction is associated with disrupted endothelial junctions, dilation of the lymphatic lumen, and lymph leakage. Proteomic analysis has shown significant dephosphorylation of ERK1/2 in meningeal lymphatic endothelial cells, correlating with the structural damage observed in MLVs. 88 Moreover, as individuals age, meningeal lymphatic drainage function declines. 86 This decline in meningeal lymphatic function has also been linked to age-related neurological conditions such as cognitive decline and neuroinflammation. Thus, the dysfunction of MLVs is both a key factor in the pathophysiology of CSDH and a consequence of aging. Reduced clearance may impair the removal of pro-inflammatory mediators and blood breakdown products from the subdural space, thereby prolonging the inflammatory and angiogenic environment that sustains hematoma growth.
Arachnoid granulations—also called Pacchioni granulations or arachnoid villi—are secretory lymphoid structures capped by multiple cell layers continuous with the dura mater and receive nerve innervation from dural connective tissue.89–93 With aging, these caps become fibrotic and thickened, potentially compromising their function. Arachnoid granulations increase in size and number in response to increased CSF pressure and are found to be highly developed in patients with chronic inflammatory conditions such as hypertension, nephritis, and atherosclerosis.89,94,95 A recent meta-analysis found that renal disease and heart disease are significant predictors of CSDH recurrence following primary surgical evacuation. 96 These findings underscore that impaired lymphatic clearance—both via MLVs and arachnoid granulations—actively contributes to CSDH. Structural vessel damage, age-related decline, and systemic comorbidities converge to reduce clearance of protein-rich, pro-inflammatory hematoma fluid. This stagnant environment promotes persistent angiogenesis, membrane thickening, and ongoing microhemorrhage, potentially explaining why elderly patients and those with cardiovascular disease often experience higher recurrence rates. 97
Matrix remodeling and meningeal reconstruction
Wound-healing macrophages play a central role in restoring tissue integrity. They promote angiogenesis, clear fibrin, and secrete MMPs—most notably MMP-2 and MMP-9.21,32,98 MMP-2, chiefly secreted by fibroblasts and macrophages, degrades gelatin and collagen types IV, V, and VII, playing a vital role in early wound healing and angiogenesis. By contrast, MMP-9, primarily released by neutrophils and macrophages, targets similar substrates but is more closely tied to inflammation, with its expression upregulated by pro-inflammatory cytokines such as IL-6 and IL-8.99,100
In CSDH, MMP-9 levels are lower in hematoma fluid than in plasma, yet they rise in recurrent CSDHs, suggesting that prolonged or excessive remodeling may contribute to recurrence. 64 The anti-inflammatory cytokine IL-10 tempers MMP-9 activity, preventing excessive extracellular matrix breakdown and guarding against further tissue injury. 64 MMP activity represents a double-edged sword—facilitating necessary vascular and connective tissue remodeling while, if unchecked, contributing to pathological membrane changes that sustain hematoma growth. The observed rise of MMP-9 in recurrent CSDH suggests that targeting inflammation-driven remodeling could be a therapeutic consideration.
Meningeal repair is also regulated by the AKAP12 scaffolding protein, which controls the epithelial–mesenchymal transition in response to cues from TGF-β1, retinoic acid, and oxygen tension.28,101 Through the TGF-β1-non-Smad-SNAI1 signaling pathway, AKAP12 enables rapid meningeal reconstruction by guiding meningeal cells to migrate toward the lesion site, thereby creating a protective barrier for the CNS. 72
Following a head injury, dural collagen synthesis continues actively. Elevated concentrations of procollagen propeptides have been detected in subdural hematoma fluid, rising rapidly within the first 2 weeks and remaining elevated for at least 3 months. 27 This indicates sustained collagen production as part of the healing process. However, aging significantly impairs this repair capacity—baseline collagenase production and cellular junction function are diminished in aged meninges. 26 Experimental models show that in young mice, collagenase production increases after injury, aiding membrane resolution. In aged mice, this response is blunted, suggesting slower or incomplete meningeal repair. Effective CSDH resolution depends on a delicate balance between extracellular matrix degradation and collagen synthesis. Younger patients mount a stronger, more coordinated remodeling response, while aging meninges—already compromised in baseline repair function—are more vulnerable to incomplete healing, persistent inflammation, and recurrence. This repair deficit in elderly patients helps explain their disproportionately higher recurrence rates.
Discussion
The pathophysiology of CSDH involves a complex interplay of inflammatory and angiogenic processes, creating a self-sustaining cycle that drives chronicity and recurrence (Figure 3). The initiating cleavage within the DBC layer sets the stage for fibroblast proliferation and collagen deposition through TGF-β1–SMAD signaling, leading to pathological membrane formation. 28 These membranes, particularly the vascularized outer layer, are characterized by fragile capillaries lacking smooth muscle and pericyte support, making them highly permeable to red blood cells and plasma. 30

Diagrammatic representation of the pathophysiological processes that contribute to the chronicity of CSDH, leading to neovascularization.
Inflammation amplifies this membrane pathology, with immune profiling revealing elevated dendritic cells and macrophages that generate ROS and cytokines. 31 OPN, which is significantly elevated in CSDH fluid, promotes leukocyte recruitment and fibroblast activation, while co-expression of OPN with VEGF in membranes amplifies integrin signaling and vessel remodeling.36,37,102 Fibroblast-derived angiogenic ligands such as CXCL12 and PGF, along with an Ang-2 predominance, destabilize new vessels. 42 Endothelial PDGF recruits pericytes to nascent vessels, but this is insufficient to establish a stable vasculature given the high VEGF expression.44,45 By linking OPN-primed fibroblast activation with VEGF-driven endothelial proliferation in a milieu of Ang-2 predominance, the membrane not only forms but progressively destabilizes its own neovasculature, predisposing to leak and rebleed.
Fibrinolysis further destabilizes the hematoma environment, as t-PA and uPAR promote plasmin generation, disrupting clot stability and activating MMPs to remodel matrix tissue.39,72 MMP-2 further remodels the extracellular matrix and releases stored VEGF, sustaining angiogenesis.40,41 Therefore, moderating local fibrinolysis could hypothetically lower free VEGF bioavailability and attenuate membrane vascularity. Hypoxia worsens this loop: mass effect reduces perfusion, stabilizing HIF-1α and upregulating VEGF, while IL-6 activation of JAK/STAT3 widens endothelial junctions and claudin-5 disruption increases vascular permeability.49,65,66,76–80,103–105
Resolution is further impaired by defective clearance. Meningeal lymphatic dysfunction, marked by disrupted endothelial junctions and lumen dilation, is strongly associated with higher recurrence.85–88 Age-related fibrosis of arachnoid granulations reduces CSF and protein clearance.89,94,95 Supporting this, β-TP, a CSF marker, is present in 93% of hematoma samples, showing that CSF entry sustains expansion and chronicity. 74 Clearance failure therefore does more than permit fluid accumulation; it prolongs the exposure of fibroblasts and endothelium to trapped cytokines and VEGF, locking the hematoma into a self-perpetuating cycle.
Taken together, recurrence persists because each pathological node reinforces the others. Fibroblast-driven membrane formation sustains leaky vasculature, inflammation and OPN-VEGF signaling accelerate angiogenesis, fibrinolysis releases more VEGF, hypoxia destabilizes junctions and upregulates VEGF, and lymphatic dysfunction traps cytokines and CSF, creating a circular self-sustaining cycle. Table 4 summarizes the key pathological events contributing to the progression of the cycle of CSDH, the agents implicated, and available therapeutic interventions.
Summary of the key pathological events contributing to the progression of the cycle of CSDH, the agents implicated, and available therapeutic interventions.

cDC2, classical dendritic cells; CSDH, chronic subdural hematoma; CSF, cerebrospinal fluid; DBC, dural border cell; HIF-1α, hypoxia-inducible factor-1alpha; MLV, meningeal lymphatic vessel; MMP, matrix metalloproteinases; NETs, neutrophil extracellular traps; OPN, osteopontin; PDGF-BB, platelet-derived growth factor receptor; PGF, placental growth factor; ROS, reactive oxygen species; TGF-β1, transforming growth factor-beta 1; tPA, tissue plasminogen activator; uPAR, urokinase plasminogen activator receptor; VEGF, vascular endothelial growth factor; βTP, β-trace protein.
Therapeutic implications follow directly from this synthesis of molecular cross-talk. Because VEGF is involved at multiple stages, it emerges as a central driver of CSDH. Targeting VEGF offers the unique advantage of disrupting several reinforcing pathogenic loops simultaneously. Bevacizumab, a VEGF inhibitor FDA-approved for the treatment of cancer, has shown promise in CSDH. Its effect was first observed incidentally in a patient treated for recurrent glioblastoma in 2016. 106 Subsequent evidence includes a 2023 case report and a 2024 case series demonstrating regression of CSDH after intra-arterial administration of bevacizumab via the MMA.67,70 An ongoing phase I/II clinical trial is evaluating the safety and efficacy of bevacizumab dosed at 2 mg/kg per affected hemisphere in approximately 140 patients with CSDH, with completion expected in 2028 (ClinicalTrials.gov identifier: NCT06510582). These early findings suggest that VEGF inhibition may represent a mechanism-driven strategy for select patients, warranting further clinical investigation.
Clinical considerations and future directions
The CSDH cycle translates into measurable clinical outcomes, with recurrence rates ranging from 10% to 30%.107,108 Predictors include preoperative seizures, postoperative midline shifting of at least 5 mm, and diabetes mellitus, though evidence for diabetes is inconsistent.107,109,110 Postoperative subdural drains reduce recurrence by facilitating the removal of residual hematoma fluid.111–113 Anticoagulation or antithrombotic therapy, traditionally viewed as high risk, may be safely resumed as early as 3 days post-treatment without significant recurrence, balancing thromboembolic and hemorrhagic risks.114,115 Given the thromboembolic risk associated with anticoagulant interruption, earlier treatment following incidental finding of CSDH—particularly with minimally invasive approaches—may warrant consideration in select patients to prevent progression to symptomatic disease requiring surgical intervention and unnecessary anticoagulant discontinuation.116–119
Surgical drainage should be performed when patients present with significant neurological symptoms, large hematoma volume, or marked midline shift, as these features indicate mass effect requiring urgent decompression. 120 MMA embolization has been increasingly adopted for CSDH, with the number of active trials rising from 2 in 2019 to 21 in 2025. 121 When combined with surgical drainage, MMA embolization reduces recurrence rates to 4.7% without increasing complications. 122 In select patients who are poor surgical candidates or have minimally symptomatic hematomas, standalone MMA embolization may be considered; meta-analytic data demonstrate a recurrence rate of 6.8%. 123 Non-target embolization remains a recognized risk, with pooled neurological complication rates of 3.8%. 124
These data highlight the importance of individualized risk stratification based on both patient factors and pathophysiological context. Incorporating insights from CSDH pathophysiology reframes management by shifting emphasis toward upstream disease drivers (Figure 4). Pharmacologic strategies targeting central mechanisms may modify disease biology, potentially reducing recurrence and improving long-term outcomes. This approach supports a multimodal treatment paradigm integrating surgical, endovascular, and drug therapies.

Summary of the key pathological events and molecular drivers contributing to the progression of the cycle of CSDH. Following initial injury and membrane formation, CSDH progression is driven by inflammation, angiogenesis, fibrinolysis with recurrent bleeding, fluid accumulation, hypoxia, lymphatic dysfunction, and matrix remodeling, forming a self-perpetuating cycle.
Conclusion
The pathophysiology of CSDH is driven by a multifactorial cascade involving persistent inflammation, disordered angiogenesis, and impaired lymphatic drainage. Advancing age exacerbates these processes, contributing to the chronicity and high recurrence rates observed in affected patients. Elucidating the underlying molecular and cellular mechanisms is essential for the development of targeted, mechanism-based therapies. As the global incidence of CSDH continues to rise, the integration of such targeted interventions into clinical practice is increasingly urgent. Continued translational research and large-scale clinical trials are critical to refining treatment strategies and establishing evidence-based guidelines for effective management.